
Abstract
Intestinal immunity has been closely associated with the pathogenesis and progression of renal diseases, a relationship known as the “gut–kidney axis.” To determine the association between immunoglobulin A nephropathy (IgAN) and Crohn’s disease (CD), a clinico-pathological study was performed on patients who had IgAN with CD (CD-IgAN) and without CD (NOS-IgAN). We enrolled 29 patients diagnosed with IgAN via renal biopsy at the Tokyo Yamate Medical Center from 2009 to 2017. The patients were divided into CD-IgAN (n = 18) and NOS-IgAN (n = 11) and evaluated for clinical and pathological findings. IgA subclasses and galactose-deficient IgA1 (Gd-IgA1) were examined via immunohistochemistry using formalin-fixed paraffin-embedded sections from renal biopsy. Our results showed no significant difference in the extent of mesangial IgA subclasses or Gd-IgA1 deposition according to the presence or absence of CD. Pathologically, however, those with CD-IgAN had remarkably higher percentage of global glomerulosclerosis and extent of interstitial fibrosis and tubular atrophy (IF/TA) compared to those with NOS-IgAN. Moreover, the extent of macrophage infiltration in the glomerulus and interstitium was significantly higher in CD-IgAN than in NOS-IgAN. Clinically, the CD-IgAN group had significantly worse responsiveness to steroid treatment compared to the NOS-IgAN group. In conclusion, the similar immunological characteristics of deposited IgA molecules in the glomeruli between the CD-IgAN and NOS-IgAN groups might suggest their etiological similarity. However, a renal pathology showing advanced glomerular and tubulointerstitial sclerosis accompanying increased macrophage infiltration and highly resistant clinical features in patients with CD-IgAN suggests that some pathophysiological factors in CD, including abnormal intestinal immunity, may promote and activate the inflammatory process in IgAN via undetermined mechanisms.
Similar content being viewed by others


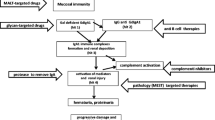
Introduction
Recent studies have highlighted the importance of the gut–kidney axis and the involvement of inflammatory bowel disease (IBD) in renal disease [1]. Several studies on renal biopsy specimens from patients with IBD have reported complications of immunoglobulin A nephropathy (IgAN) [2, 3].
A study of 83 patients with IBD by Josephine et al. showed that IgAN was the most frequent complication (20 cases) [4]. Other studies have also investigated the relationship between CD and IgAN [5, 6]. Immunoglobulin A (IgA) has been suggested to play a major role in intestinal immunity because it is produced by IgA-producing B/plasma cells and secreted into the intestinal lumen through the intestinal epithelium. By contrast, studies have suggested that IBD, such as Crohn’s disease (CD) and ulcerative colitis, which have been associated with intestinal immune disorders, impairs IgA production and secretion. However, the precise B cell abnormalities underlying IgAN have yet to be elucidated [7]. Based on the aforementioned findings, the US NEFIGAN study reported that budesonide, a CD therapeutic and intestinal steroid, can be used to treat patients with IgAN and ameliorated proteinuria [8] [9]. Moreover, another study showed that budesonide was effective for IgAN that recurred after kidney transplantation [10]. Budesonide targets the Peyer’s patches of the intestine and is thought to suppress abnormal T-cell immunity, suggesting that modifying the intestinal immunity might result in renal disease.
IgAN, which is characterized by the deposition of IgAN immune complex in the mesangial region of the glomerulus, can be classified as idiopathic IgAN or secondary IgAN associated with infectious and liver diseases. In patients with idiopathic IgAN, the galactose-deficient IgA1 (Gd-IgA1) has a structure in which galactose is deficient in the O-linked sugar chain at IgA1 hinge and N-acetyl galactosamine (GalNAC) is exposed. Reports have shown that IgG immune complexes against this abnormal IgA1 are deposited in the glomerulus [11] [12]. In fact, the IgA subclass deposited in the glomerulus of most patients with idiopathic IgAN mainly include IgA1, with the quantity of IgA2 co-deposition being negligible. The increased level of serum Gd-IgA1 in IgAN [13], as well as the significant correlation between the extent of mesangial Gd-IgA1 deposition and clinical disease activity of IgAN, suggests the etiological role of Gd-IgA1 in IgAN [14]. However, recent evidence has shown that secondary IgAN, which occurs in various disease, can also be attributed to the deposition of Gd-IgA1 in the mesangial region [15]. Therefore, Gd-IgA1 may not be specific to primary IgAN, suggesting a similar immunological mechanism between secondary and primary IgAN [16].
To elucidate the pathogenetic relationship between IgAN and CD, we compared the clinical and histological findings of patients diagnosed with CD prior to IgAN (CD-IgAN) and those who had IgAN without CD (NOS-IgAN). In particular, we analyzed the IgA subclass composition, which is thought to be involved in the pathogenic mechanism of IgAN, the presence or absence of Gd-IgA1, and the degree of deposition.
Materials and methods
Patients and study design
We enrolled 29 adult patients (≥ 18 years old) who were diagnosed with IgAN based on the findings of renal biopsy specimens obtained between January 2009 and December 2017 at Tokyo Yamate Medical Center. The included patients were categorized into the following two groups: (1) CD-IgAN (n = 18) and (2) NOS-IgA (n = 11). Consecutive patients in the NOS-IgAN group were selected during the same period when patients in the CD-IgAN group were enrolled. The patients were not age or sex matched when selecting the two study groups. No selection bias was implemented during the enrollment to the NOS-IgAN group. The higher number of patients with CD-IgAN than those with NOS-IgAN was because our hospital specializes in treatment of inflammatory bowel diseases, and the male predominance in CD-IgAN group was caused by the male predominance of IBD in our hospital. The clinical and histopathological characteristics were examined.
IgAN was pathologically diagnosed through glomerular IgA deposition and C3 deposition determined via immunofluorescent microscopy (IF). Moreover, clinical information was collected. Patients whose renal biopsy specimens had insufficient sample volume or contained no glomerulus were excluded from the study. When more than one renal biopsy was performed during the observation period, the first biopsy sample was selected for histological evaluation.
For the diagnosis of CD, we included individuals whose diagnosis was confirmed by our gastroenterologists based on clinical, endoscopic, and histopathological findings of colorectal specimens before the renal biopsy.
All patients provided informed consent at the time of renal biopsy and were able to opt-out from the study if they desired. This study was approved by the Ethics Committee of the Tokyo Yamate Medical Center (J023) and conducted in accordance with the ethical standards enshrined in the Declaration of Helsinki.
Clinical parameters
The following variables patient characteristics were retrospectively obtained from the medical records: age, gender, duration from the disease onset to renal biopsy (years), body mass index (BMI) (kg/m2), body weight (kg), systolic blood pressure (mmHg), use of renin–angiotensin system inhibitors (RASI), serum creatinine (Cr) level (mg/dL), serum IgA level (mg/dL), hematuria, and proteinuria. Hematuria and proteinuria (before and after treatment) were scored semi-quantitatively based on the urine test strip method. The grade was determined as 0, 0.5, 1, 2, and 3 when the result was ( −), ( ±), (1 +), (2 +), and (3 +) on the strip test, respectively. Treatments for IgAN were divided into oral steroids, intravenous steroid pulse (IVP), tonsillectomy, and their combination, such as oral steroid therapy and tonsillectomy. The number of patients administered 5-aminosalicylic acid (5-ASA) was only counted in those with CD-IgAN. To evaluate therapeutic response, we compared the number of patients with hematuria (score ≥ 1) and proteinuria (score ≥ 1) before and after the treatment in the CD-IgAN and NOS-IgAN groups.
Histopathological assessment
Histopathologic findings were independently reviewed by a renal pathologist (K.H.) and nephrologist (M.A.) who were blinded to the clinical background of each section. The rate of global glomerular sclerosis was defined as the percentage of globally sclerotic glomerulus among all glomeruli in one section. The rate of active phase crescent was defined as the rate of cellular or fibrocellular crescent among all glomeruli in one section. Furthermore, we evaluated the histological scoring, MEST-C, using the Oxford’s classification for IgAN [17, 18].
We performed periodic acid–Schiff stain and Masson trichrome stain (Masson) and classified the extent of interstitial fibrosis and tubular atrophy (IF/TA) into five grades according to the percentage of affected area in the total cortical area: 0 (0–5%), 1 (6–25%), 2 (26–50%), 3 (51–75%), and 4 (76–100%). The extent of interstitial inflammatory cell infiltration was also evaluated and semi-quantitatively classified into four grades: 0 (absent), 1 (mild), 2 (moderate), and 3 (severe). Vascular lesions were evaluated based on the degree of intimal hyalinosis and luminal narrowing in the arterioles and degree of intimal thickening accompanied by the proliferation of elastic fibers and luminal narrowing in the interlobular arteries. Arteriolar hyalinosis and interlobular arteriosclerosis were graded as 0 (absent intimal lesion), 1 (mild intimal lesion without luminal narrowing), 2 (moderate intimal lesion with unremarkable luminal narrowing), and 3 (severe intimal lesion with remarkable luminal narrowing) depending on the severity of each lesion.
To compensate for the small cohort size of the NOS-IgAN group in this study, a meta-analytic comparison of MEST-C scores was performed using two large-scale IgAN cohorts included in previous studies in the literature [19, 20].
Immunohistological assessment
The staining protocol for Gd-IgA1 was described in the previous literature [14], and glomerular Gd-IgA1 deposition was examined via immunohistochemistry (IHC). Briefly, deparaffined sections were heated with buffer (Histofine®, Nichirei, Tokyo, Japan) in an autoclave at 121 °C for 30 min for antigen retrieval. After endogenous peroxidase was quenched with 0.3% H2O2 in methanol, nonspecific binding was blocked using protein blocking solution, and the sections were incubated overnight at 4 °C with rat monoclonal anti-human Gd-IgA1 antibody (KM55) diluted to 1:100, followed by EnVision™ + Dual Link System-HRP (Dako, Glostrup, Denmark) for 60 min at room temperature. Diaminobenzidine (DAB) (Dako) was used for visualization.
The staining intensity of IgA (#AR045-5R, BioGenex), IgA1(#9130–01, Southern Biotech, 1:20,000 antibody dilution, activation time 15 min), IgA2 (#9140–01, Southern Biotech,1:10,000 antibody dilution, activation time 15 min), and Gd-IgA1 (KM55, #10,777, Immuno-Biological Laboratories) in each glomerulus was classified into four grades: 0 (negative), 1 (weak, 1 +), 2 (moderate, 2 +), and 3 (strong, 3 +). One pathological slide was assigned to each case, and all glomeruli in one slide were classified into four grades. All of glomeruli on each slide were assessed and categorized into four grades. Thereafter, staining intensity was determined as the summation of these grades divided by the total number of glomeruli on each slide. Immunofluorescent intensity of glomerular C3 deposition was evaluated and classified into four grades: 0 (negative), 1 (weak, 1 +), 2 (moderate, 2 +), and 3 (strong, 3 +) based on the descriptions in laboratory reports of routine immunofluorescent examination using frozen sections of the specimens.
We also evaluated glomerular and interstitial macrophage infiltration by immunohistochemistry using primary monoclonal antibody against CD68 (clone: PG-M1, DAKO, Glostrup, Denmark). For glomerular macrophage infiltration, the extent of glomerular CD68 ( +) cell infiltration was evaluated semi-quantitatively in the microscopic field at high magnification (× 400) and classified into four grades: 0 (none), 1 (mild, < 5 cells/glomerulus), 2 (moderate, 5–9 cells/glomerulus), and 3 (severe, ≥ 10 cells/glomerulus). For interstitial macrophage infiltration, the extent of interstitial CD68 ( +) cell infiltration was semi-quantitatively evaluated in the microscopic field at the middle magnification (× 200) and classified into four grades: 0 (none), 1 (mild), 2 (moderate), and 3 (severe).
Statistical analysis
Data were expressed as the means ± SD or percentages (%). Nonparametric continuous variables were compared using Mann–Whitney U test. Categorical variables were compared using Fisher’s test and McNemar’s test. Meta-analytic comparison of the MEST-C scores using the cohorts included in previous literatures was performed using Fisher’s exact probability test. We also evaluated the correlation between disease duration of CD and IF/TA grade or global glomerular sclerosis rate using Spearman’s rank correlation coefficient and created scatter plots. All statistical analyses were performed using EZR (Saitama Medical Center, Jichi Medical University, Saitama, Japan), a graphical user interface for R (The R Foundation for Statistical Computing, Vienna, Austria), with p values < 0.05 indicating statistical significance.
Results
Clinical characteristics of patients
Table S1 shows the clinical background and laboratory findings of the 29 patients with IgAN studied, among whom 18 and 11 belonged to the CD-IgAN and NOS-IgAN groups, respectively. The mean serum creatinine level at renal biopsy was 1.5 (± 0.8) and 0.9 (± 0.3) mg/dL for the CD-IgAN and NOS-IgAN groups, respectively, with the former having significantly higher levels (p = 0.003). BMI was 20.8 (± 3.0) and 23.8 (± 3.3) kg/m2 for CD-IgAN and NOS-IgAN, respectively, with the former having significantly lower mean values (p = 0.012). Regarding sex, 17 (94.4%) and 6 (54.5%) subjects in the CD-IgAN and NOS-IgAN groups were males, with the former being more male predominant (p = 0.018). Aside from gender, serum creatinine level, and BMI, no significant differences between the two groups were noted in the following clinical backgrounds: duration from onset of urinary abnormality to biopsy, blood pressure, hematuria or proteinuria grade, serum IgA level, and therapeutic intervention for IgAN.
Comparison of histological findings between the CD-IgAN and NOS-IgAN groups ( Table 1 )
Figure 1 shows the typical renal biopsy findings for the glomerulus and tubulointerstitium in the CD-IgAN and NOS-IgAN groups. Glomerular and interstitial macrophage infiltrations detected by anti-CD68 immunohistochemical staining are also presented. Cases with CD-IgAN had more prominent sclerosing glomerular lesions compared to those with NOS-IgAN. CD-IgAN had more extensive and severe fibrosis than NOS-IgAN. Glomerular and interstitial macrophage infiltrations were observed in both patients depending on glomerular and interstitial lesions.

Typical histological findings and macrophage infiltration of the glomerulus and tubulointerstitium in the CD-IgAN and NOS-IgAN groups. The upper row shows the staining findings of CD-IgAN (A, B, and C) and the lower row shows the staining findings of NOS-IgAN (D, E, and F). Glomerular sclerosis and IF/TA are more severe in CD-IgAN than in NOS-IgAN. Glomerular and interstitial macrophage infiltration was observed with various degrees in both groups, depending on the histological alterations. (A and D, periodic acid–Schiff stain; B and E, Masson trichrome stain; C and F, CD68 immunohistochemical stain, bar = 50 μm)
The differences in histopathological findings between the 18 CD-IgAN cases and 11 NOS-IgAN cases are quantified and compared in Table 1. The mean global glomerular sclerosis rate was 29.6% (± 31.3) and 5.1% (± 6.0) in the CD-IgAN and NOS-IgAN groups, with the former showing a significantly higher rate (p = 0.023). The mean proportion of glomeruli containing active crescents was 7.5% (± 10.7) and 1.9% (± 3.4) for the CD-IgAN and NOS-IgAN groups, with no significant differences between both groups (p = 0.22), whereas in patients subsequently treated with steroids, it was significantly higher in the CD-IgA group (n = 9) than in the NOS-IgAN group (n = 9) (12.3% [± 11.1] and 2.3% [± 3.7], respectively [p = 0.02]). Among the MEST-C scores defined via the Oxford classification, T score was significantly higher in CD-IgAN group than in NOS-IgAN group (0.56 ± 0.71, 0 ± 0, p = 0.046), whereas the other scores including M, E, S, and C did not show any significant differences between the CD-IgAN and NOS-IgAN groups.
Concerning tubulo-interstitial lesions, none of the patients with NOS-IgA showed interstitial fibrosis/tubular atrophy (IF/TA) grade 2 or higher lesions, whereas 9 out of 18 cases (50%) with CD-IgAN had grade 2 or higher lesions, suggesting that the CD-IgA group had significantly higher IF/TA grade compared to the NOS-IgAN group (1.50 ± 0.99 vs. 0.36 ± 0.50, p = 0.017), whereas the extent of interstitial inflammatory cell infiltration tended to be higher in the CD-IgAN group than in the NOS-IgAN group but not significantly different (1.17 ± 0.62 vs. 0.64 ± 0.50, p = 0.092).
The comparison of vascular lesions yielded the following results. Arteriolar hyalinosis was observed in 44% of cases with CD-IgAN but none of those with NOS-IgAN (p = 0.012). Moreover, interlobular arteriosclerosis was detected in 21% of the patients with CD-IgAN and but none of those with NOS-IgAN (p = 0.273), although the difference was not statistically significant. The average grade of arteriolar hyalinosis was 0.61 ± 0.61 and 0 ± 0 in the CD-IgAN and NOS-IgAN groups, with the former showing a significantly higher grate (p = 0.003). However, the average grade of interlobular arteriosclerosis was not different between CD-IgAN and NOS-IgAN groups.
The extent of glomerular macrophage infiltration was significantly higher in CD-IgAN group than in NOS-IgAN group (1.45 ± 0.51 vs. 1.13 ± 0.83, p = 0.02). The extent of interstitial macrophage infiltration was also significantly higher in CD-IgAN group than in NOS-IgAN group (2.06 ± 0.68 vs. 1.13 ± 0.35, p = 0.011).
Meta-analytic comparisons of the MEST-C scores between the CD-IgAN group of the present study and the IgAN cohorts of the previous studies [19, 20] revealed that the incidence of T1/T2 was significantly higher in our CD-IgAN group than in Barbour’s IgAN cohort [19] (44.4% vs. 22.1%, p = 0.045) and Kamano’s cohort [20] (44.4% vs. 11.4%, p < 0.001). Conversely, the incidence of S1 was significantly lower in our CD-IgAN group than in Barbour’s IgAN cohort (22.2% vs. 75.0%, p < 0.001) and Kamano’s cohort (22.2% vs. 79.0%, p < 0.001). Additionally, the incidences of E1 and C1/2 in our CD-IgAN group were significantly lower than those in Kamano’s cohort (E1: 5.6% vs. 35.3%, p = 0.0098, C1/2: 33.3% vs. 59.1%, p = 0.005) but did not significantly differ from those in Barbour’s cohort (Table S3). Furthermore, the comparisons of MEST-C scores between our NOS-IgA group and these two cohorts revealed that the incidence of S1 was significantly lower in our NOS-IgA group than in both cohorts (0% vs.75.0% and 79.0%, p < 0.001). The incidences of E1 and C1/2 in our NOS-IgA group were also significantly lower than those in Kamano’s cohorts (E1: 0% vs. 35.3%, p = 0.011 and C1/2: 18.2% vs. 59.1%, p = 0.010) but did not differ from those in Barbour’s cohort. No significant difference in M and T scores was observed between our NOS-IgAN cohort and the two large-scale IgAN cohorts (Table S4).
No significant correlation between Crohn’s disease duration and global glomerular sclerosis or IF/TA
Figure S1 shows the correlation between CD duration and global glomerular sclerosis (A) or IF/TA (B). No significant correlation was observed between global glomerular sclerosis and the duration of CD (Spearman’s rank correlation coefficient 0.39, p = 0.11). Similarly, no significant correlation was observed between IF/TA grade and the duration of CD (Spearman’s rank correlation coefficient 0.45, p = 0.060). Moreover, the extents of arteriolar hyalinosis and interlobular arteriosclerosis were not correlated with the duration of CD (data not shown).
Comparison of immunostaining results for IgA, C3, IgA subclass, and Gd-IgA1 between the CD-IgAN and NOS-IgAN groups
IgA subclass comprises IgA1 and IgA2, and representative image of each staining grade is shown in Figure S2. IgA1 and IgA2 were respectively deposited specifically in the mesangial region. No difference in deposition site of IgA subclass was observed regardless of the presence or absence of CD. The extent of IgA, IgA1, IgA2, and Gd-IgA1 deposition was represented as the average staining grade of all glomeruli on each specimen and compared between the CD-IgAN and NOS-IgAN groups (Table 2). Although the staining intensity of IgA, IgA1, and IgA2 was generally higher in NOS-IgAN than in CD-IgAN, a statistical significance was demonstrated only in IgA1 (1.83 ± 0.86 vs. 2.66 ± 0.80, p = 0.029) but not in IgA (2.26 ± 0.67 vs. 2.49 ± 0.42, p = 0.52) and IgA2 (1.39 ± 0.86 vs. 1.77 ± 0.26, p = 0.155) according to the presence and absence of CD complication. Moreover, no significant difference in the intensity ratio of IgA2/IgA1 was found between the two groups (0.78 ± 0.27 vs. 0.70 ± 0.26, p = 0.469).
Thereafter, the typical glomerular depositions of Gd-IgA1 staining were assessed separately for the CD-IgAN and NOS-IgAN groups (Fig. 2). Gd-IgA1 was specifically deposited in the mesangial region, with no significant difference according to the presence and absence of CD complication, similar to the IgA subclass. The extent of Gd-IgA1 deposition was evaluated by similar manner performed in IgA subclass evaluation and compared between the CD-IgAN and the NOS-IgAN groups (Table 2). No significant difference in the staining intensity was observed according to the presence or absence of CD complication (1.17 ± 0.79 vs.1.52 ± 0.65, p = 0.240). The grade of C3 deposition was semi-quantitatively evaluated by routine immunofluorescent examination for renal biopsy diagnosis. Although 3 of the 14 CD-IgAN patients (21.4%) and none of the 9 NOS-IgAN patients (0%) were negative for C3 deposition, the mean intensity grade of C3 deposition was not different statistically between the CD-IgAN and the NOS-IgAN groups (1.57 ± 1.02 vs. 2.22 ± 0.67, p = 0.543).

Typical Gd-IgA1 staining in glomeruli in the CD-IgAN and NOS-IgAN groups. The upper row shows the staining findings of CD-IgAN (A, B, C, and D), and the lower row shows the staining findings of NOS-IgAN (E, F, G, and H). From left to right, glomeruli with negative, + 1, + 2, and + 3 staining intensities are shown (× 400; bar = 50 μm). There was no difference in the deposition site and morphology of Gd-IgA1 due to the presence or absence of CD
Comparison of steroid treatment effects between the CD-IgAN and NOS-IgAN groups
Figure S3 presents the changes in hematuria and proteinuria grade before and after steroid treatment only for targeting treated cases classified according to the presence or absence of CD. In the NOS-IgAN group, treatment significantly ameliorated both hematuria and proteinuria (p = 0.041 and 0.041, respectively). In the CD-IgAN group, however, poor amelioration of both hematuria and proteinuria was observed.
Steroid treatment was performed in nine cases with CD-IgAN (50%) and nine cases with NOS-IgAN (91%). Table S3 details the number of cases for each hematuria and proteinuria grade before and after treatment in both groups described in Figure S3. Among cases with CD-IgAN, 22.2% retained a hematuria grade of 3 + , whereas 22.2% retained a proteinuria grade of 3 + even after treatment. However, none of the NOS-IgAN cases without CD had a hematuria or proteinuria grade of 3 + after steroid treatment (Fig. 3).

Histological grading of glomerular and interstitial macrophage infiltration. The upper row shows the CD68 staining findings of glomerulus (A, B, C, and D), and the lower row shows the CD68 staining findings of tubulointerstitium (E, F, and G). From left to right, glomerular and interstitial CD68 ( +) cell infiltrations with grades 0, 1, 2, and 3 of staining intensities are shown. Glomerular macrophage infiltration was classified into four grades: 0 (none), 1 (mild, under 5 cells/glomerulus), 2 (moderate, 5–9 cells/glomerulus), and 3 (severe, 10 and over cells/glomerulus). Interstitial CD68 ( +) cell infiltration was classified into four grades: 0 (none), 1 (mild, under 10 cells/field at × 200), 2 (moderate, 10–29 cells/field at × 200), and 3 (severe, 30 and over cells/field at × 200). There was no interstitial area with grade 0 CD68 staining (absent for macrophage infiltration) in all specimens (CD68 immunohistochemical stain, × 400 in upper row and × 200 in upper row; bar = 50 μm; N/A, not available)
Discussion
IBD may be complicated with IgAN [21, 22], with studies suggesting an association between intestinal immunity and the pathogenesis of IgAN. Therefore, the current study investigated the clinical and pathological differences between IgAN associated with IBD and general IgAN associated with upper respiratory tract inflammation, such as tonsillitis. The plasma cells of the upper respiratory tract mucosa primarily produces IgA1 [23]. However, the intestinal mucosa, especially Peyer’s patches, has been consider to predominantly secrete IgA2 (approximately 60% in mucosal cells) instead of IgA1 [24]. In the case of IgAN with CD, intestinal IgA2 may have been deposited in the glomerular mesangium and involved in the induction and progression of IgAN. However, our results indicated that among the deposited IgA subclasses, the IgA1 subclass was predominant in both the CD-IgAN and NOS-IgAN groups, with no significant difference in staining strength of IgA2 between both groups.
Thereafter, we investigated the deposition of Gd-IgA1, which has been reported to be specifically deposited in the glomeruli of primary IgAN [25]. Gd-IgA1 is an abnormal IgA1 of the IgA1 subclass that exhibits a structure in which galactose is deficient in the o-linked sugar chain at the hinge and N-acetyl galactosamine (GalNAC) is exposed [11, 12]. Notably, the current study found no difference in the degree of Gd-IgA1 deposition in the glomeruli regardless of CD complications, suggesting no significant difference between the CD-IgAN and NOS-IgAN groups in terms of the deposition of the Gd-IgA1 complex. In recent years, Gd-IgA1 has been detected in both secondary and primary IgAN, suggesting that secondary IgAN shares the same pathogenesis as primary IgAN. Additionally, negative views have emerged regarding the disease specificity of Gd-IgA1 [26].
After analyzing the histological findings of the kidney, the current study found that cases of CD-IgAN had significantly more severe global glomerulosclerosis, arteriolar hyalinosis grade, and IF/TA than those with NOS-IgAN. Comparing the Oxford classification scores revealed that the T score representing IF/TA was significantly higher in the CD-IgAN group than in the NOS-IgAN group. To confirm this tendency, we performed a meta-analytic comparison of MEST-C score using large-scale cohorts of IgAN patients reported in the previous literatures. The incidence of T1/2 was higher in our CD-IgAN group than in the large-scale IgAN cohorts reported by Barbour et al. from Canada [19] and Kamano et al. from Japan [20].
We speculate that these histological differences indicating advanced glomerulosclerosis and tubulointerstitial changes observed in the CD-IgAN patients were associated with the following three factors: (1) pathophysiology of CD (i.e., diarrhea and dehydration), (2) therapeutic agents for CD, and (3) systemic inflammation, including the intestinal tract.
First, during the course of IBD, dehydration due to diarrhea low dietary and water intake, surgery, etc. may reduce the circulating blood volume, causing tubular interstitial disorders and glomerulosclerosis [27]. Moreover, reports have showed that undernutrition and hypokalemia cause chronic tubular interstitial disorders. The reason for this is that reduced effective circulating blood volume may stimulate the renin–angiotensin–aldosterone system, followed by enhanced angiotensin II activation, causing arteriolar contraction, and glomerular ischemia, as well as interstitial fibrosis [28, 29]. In addition, evidence has shown that hyperuricemia, which is common in CD, may exacerbate glomerular sclerosis [30].
The second factor involves the effect of the therapeutics. Renal disorders associated with CD are present in 4–23% of patients with CD [31]. 5-ASA remains the main therapeutic agent for CD, with its renal adverse effects being collectively referred to as mesalamine-related kidney disease [32]. The mechanism through which mesalamine promotes renal damage appears to be through salicylate inhibition of the synthesis of intrarenal prostaglandins, which are vasoactive mediators of intrarenal blood flow and uncouple oxidative phosphorylation in mitochondria [27, 33]. Moreover, some reports have shown that mesalamine promotes renal damage histologically through interstitial nephritis [34], although similar findings having been reported in patients with CD not using 5-ASA [35]. Therefore, it is difficult to distinguish whether interstitial nephritis could be attributed to the drug or CD itself [31]. Given that 5-ASA agents were used in 12 of the 16 cases (75.0%, 2 cases not available) in this study, the advanced tubulointerstitial lesion may have been caused by the drug, although we could not precisely determine the etiology.
Third, the pathophysiology of CD itself, that is, immune abnormalities, may be involved. It is well known that macrophages and T cells produce large amounts of IL-23 and TNF-α in immune disorders, such as CD, and are considered to play a central role in the pathophysiology of CD [36]. These cytokines are also known to contribute to the exacerbation of tubulointerstitial lesions in IgAN [36]. Additionally, the mechanism by which dysfunctional macrophages promote intestinal fibrosis [37] has also been reported. In recent years, the mechanism and systemic response of B cell immune abnormalities [38, 39] have been clarified in CD [7]. B cell immune dysfunction has been reported to be involved in interstitial inflammation of chronic kidney disease, including IgAN [40], and immunological dysfunction of CD has been associated with IgAN from the viewpoint of immune dysfunction, which might act as an exacerbating factor for renal tubular interstitial disorders. Our result demonstrating the increased glomerular and interstitial macrophage infiltration in the CD-IgAN group than in the NOS-IgAN group suggests that some immunological abnormalities in CD may affect the macrophage infiltration in the kidney and promote glomerulosclerosis and interstitial fibrosis. Furthermore, in the pathology of IgAN, complement activity has been considered to promote glomerular sclerosis and interstitial fibrosis [41, 42]. In patients with CD, a previous study has shown that activated complement (mainly C3b) is strongly stained in the intestinal mucosa [43] and that the expression of complement C3 mRNA is increased in the resected ileocecal specimens [44]. Although we could not detect the difference of glomerular C3 staining intensity between CD-IgAN and NOS-IgAN, the effect of complement activation associated with CD may affect glomerular and tubulointerstitial inflammation of IgA nephropathy.
Factors considered to have caused the difference in therapeutic response to steroid therapy between the CD-IgAN and NOS-IgAN groups remain unclear. Regarding glomerulosclerosis and IF/TA, cases with IgAN who had more severe pathological changes were reported to be more resistant to steroid treatment than those with milder diseases [45]. The advanced IF/TA may be one of the factors influencing the poor therapeutic response in our CD-IgAN group; however, other factors, such as disease duration, effects of drugs, and immunological background, need to be further investigated to clarify the clinical features of IgAN complicated with CD.
The meta-analytic comparisons of MEST-C scores with two large-scale IgAN cohorts revealed several concerns related to the pathological relationship between CD and IgAN. One such concern is the lower incidence of segmental glomerulosclerosis, represented by the S score, in the CD-IgAN group than in the large-scale IgAN cohorts. We cannot speculate any inhibitory effects of CD on the formation of segmental sclerosis. As such, we believe that this tendency might have been an institutional bias on histological evaluation and the definition of segmental glomerulosclerosis in this study considering that the same tendency was observed in our NOS-IgAN group. Concerning the E and C scores, both our CD-IgAN and NOS-IgAN groups presented lower degrees of E and C compared to Kamano’s cohort but not Barbour’s cohort. Furthermore, it was our understanding that this could have been attributed to the institutional bias on histological evaluation of the E and C scores in Kamano’s study, as well as in the evaluation of the S score in the current study.
Limitations of the present study
This study has several notable limitations. First, this was a retrospective case–control analysis performed at a single center with a relatively small sample size, especially in NOS-IgAN, and with a specialized bias for patients with IBD in our hospital. Second, the treatment protocol and treatment period were not standardized. Third, there may have been problems with the detection sensitivity of the immunohistological examinations using the formalin-fixed, paraffin-embedded section instead of the frozen section. Finally, in some cases, steroids were used for CD prior to IgAN treatment, and the effects of therapeutic drugs for CD on IgA disease activity and histological findings cannot be ruled out.
Conclusions
IgA associated with CD did not differ from usual IgAN in terms of Gd-IgA1 staining and IgA subclasses in kidney biopsy specimens. These findings suggest no difference in the etiology between the CD-IgAN and NOS-IgAN groups. However, histological findings showed that patients with CD had severe glomerular sclerosis and IF/TA accompanying increased glomerular and interstitial macrophage infiltration and highly resistant clinical response to steroid treatment suggests that the immunological abnormality of CD may promote and activate the inflammatory processes of IgAN. It is necessary to accumulate further cases to clarify the relationship between CD and IgAN.
References
Evenepoel P, Poesen R, Meijers B (2017) The gut-kidney axis. Pediatr Nephrol 32:2005–2014. https://doi.org/10.1007/s00467-016-3527-x
Pohjonen J, Nurmi R, Metso M, Oksanen P, Huhtala H, Porsti I, Mustonen J, Kaukinen K, Makela S (2019) Inflammatory bowel disease in patients undergoing renal biopsies. Clin Kidney J 12:645–651. https://doi.org/10.1093/ckj/sfz004
Elaziz MMA, Fayed A (2018) Patterns of renal involvement in a cohort of patients with inflammatory bowel disease in Egypt. Acta Gastroenterol Belg 81:381–385
Ambruzs JM, Walker PD, Larsen CP (2014) The histopathologic spectrum of kidney biopsies in patients with inflammatory bowel disease. Clin J Am Soc Nephrol 9:265–270. https://doi.org/10.2215/CJN.04660513
Pouria S, Barratt J (2008) Secondary IgA nephropathy. Semin Nephrol 28:27–37. https://doi.org/10.1016/j.semnephrol.2007.10.004
Pipili C, Michopoulos S, Sotiropoulou M, Mpakirtzi T, Grapsa E (2012) Is there any association between IgA nephropathy, Crohn’s disease and Helicobacter pylori infection? Ren Fail 34:506–509. https://doi.org/10.3109/0886022X.2011.653774
Brandtzaeg P, Carlsen HS, Halstensen TS (2006) The B-cell system in inflammatory bowel disease. Adv Exp Med Biol 579:149–167. https://doi.org/10.1007/0-387-33778-4_10
Fellstrom BC, Barratt J, Cook H, Coppo R, Feehally J, de Fijter JW, Floege J, Hetzel G, Jardine AG, Locatelli F, Maes BD, Mercer A, Ortiz F, Praga M, Sorensen SS, Tesar V, Del Vecchio L, Investigators NT (2017) Targeted-release budesonide versus placebo in patients with IgA nephropathy (NEFIGAN): a double-blind, randomised, placebo-controlled phase 2b trial. Lancet 389:2117–2127. https://doi.org/10.1016/S0140-6736(17)30550-0
Coppo R (2017) Corticosteroids in IgA nephropathy: lessons from recent studies. J Am Soc Nephrol 28:25–33. https://doi.org/10.1681/ASN.2016060647
Lingaraj U, Aralapuram K, Chikkanayakanhalli S, Vishwanathan A, Vankalakunti M (2020) Successful treatment of a patient with posttransplant IgA nephropathy with targeted release formulation of budesonide. Saudi J Kidney Dis Transpl 31:521–523. https://doi.org/10.4103/1319-2442.284029
Tomana M, Matousovic K, Julian BA, Radl J, Konecny K, Mestecky J (1997) Galactose-deficient IgA1 in sera of IgA nephropathy patients is present in complexes with IgG. Kidney Int 52:509–516. https://doi.org/10.1038/ki.1997.361
Tomana M, Novak J, Julian BA, Matousovic K, Konecny K, Mestecky J (1999) Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J Clin Invest 104:73–81. https://doi.org/10.1172/JCI5535
Placzek WJ, Yanagawa H, Makita Y, Renfrow MB, Julian BA, Rizk DV, Suzuki Y, Novak J, Suzuki H (2018) Serum galactose-deficient-IgA1 and IgG autoantibodies correlate in patients with IgA nephropathy. PLoS ONE 13:e0190967. https://doi.org/10.1371/journal.pone.0190967
Wada Y, Matsumoto K, Suzuki T, Saito T, Kanazawa N, Tachibana S, Iseri K, Sugiyama M, Iyoda M, Shibata T (2018) Clinical significance of serum and mesangial galactose-deficient IgA1 in patients with IgA nephropathy. PLoS One 13:e0206865. https://doi.org/10.1371/journal.pone.0206865
Cassol CA, Bott C, Nadasdy GM, Alberton V, Malvar A, Nagaraja HN, Nadasdy T, Rovin BH, Satoskar AA (2019) Immunostaining for galactose-deficient immunoglobulin A is not specific for primary immunoglobulin A nephropathy. Nephrol Dial Transplant. https://doi.org/10.1093/ndt/gfz152
Wang M, Lv J, Zhang X, Chen P, Zhao M, Zhang H (2020) Secondary IgA nephropathy shares the same immune features with primary IgA nephropathy. Kidney Int Rep 5:165–172. https://doi.org/10.1016/j.ekir.2019.10.012
Working Group of the International Ig ANN, the Renal Pathology S, Roberts IS, Cook HT, Troyanov S, Alpers CE, Amore A, Barratt J, Berthoux F, Bonsib S, Bruijn JA, Cattran DC, Coppo R, D’Agati V, D’Amico G, Emancipator S, Emma F, Feehally J, Ferrario F, Fervenza FC, Florquin S, Fogo A, Geddes CC, Groene HJ, Haas M, Herzenberg AM, Hill PA, Hogg RJ, Hsu SI, Jennette JC, Joh K, Julian BA, Kawamura T, Lai FM, Li LS, Li PK, Liu ZH, Mackinnon B, Mezzano S, Schena FP, Tomino Y, Walker PD, Wang H, Weening JJ, Yoshikawa N, Zhang H (2009) The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility. Kidney Int 76:546–556. https://doi.org/10.1038/ki.2009.168
Trimarchi H, Barratt J, Cattran DC, Cook HT, Coppo R, Haas M, Liu ZH, Roberts IS, Yuzawa Y, Zhang H, Feehally J, Ig ANCWGotIINN, the Renal Pathology S, Conference P (2017) Oxford classification of IgA nephropathy 2016: an update from the IgA Nephropathy Classification Working Group. Kidney Int 91:1014–1021. https://doi.org/10.1016/j.kint.2017.02.003
Barbour SJ, Espino-Hernandez G, Reich HN, Coppo R, Roberts IS, Feehally J, Herzenberg AM, Cattran DC, Oxford Derivation NAV, Consortia V, American ODN, V, Consortia V, (2016) The MEST score provides earlier risk prediction in lgA nephropathy. Kidney Int 89:167–175. https://doi.org/10.1038/ki.2015.322
Kamano C, Shimizu A, Joh K, Hashiguchi A, Hisano S, Katafuchi R, Kawamura T, AnpcSG JI (2021) A cross-sectional study in patients with IgA nephropathy of correlations between clinical data and pathological findings at the time of renal biopsy: a Japanese prospective cohort study. Clin Exp Nephrol 25:509–521. https://doi.org/10.1007/s10157-021-02022-x
Filiopoulos V, Trompouki S, Hadjiyannakos D, Paraskevakou H, Kamperoglou D, Vlassopoulos D (2010) IgA nephropathy in association with Crohn’s disease: a case report and brief review of the literature. Ren Fail 32:523–527. https://doi.org/10.3109/08860221003710554
Terasaka T, Uchida HA, Umebayashi R, Tsukamoto K, Tanaka K, Kitagawa M, Sugiyama H, Tanioka H, Wada J (2016) The possible involvement of intestine-derived IgA1: a case of IgA nephropathy associated with Crohn’s disease. BMC Nephrol 17:122. https://doi.org/10.1186/s12882-016-0344-1
Perse M, Veceric-Haler Z (2019) The role of IgA in the pathogenesis of IgA nephropathy. Int J Mol Sci 20. https://doi.org/10.3390/ijms20246199
Chiba M, Ohta H, Yagisawa H, Masamune O (1987) IgA1 & IgA2 distribution in the intestine. Gastroenterol Jpn 22:18–23
Martin-Penagos L, Fernandez-Fresnedo G, Benito-Hernandez A, Mazon J, de Cos M, Oviedo MV, San Segundo D, Lopez-Hoyos M, Gomez-Roman J, Ruiz JC, Rodrigo E (2021) Measurement of galactosyl-deficient IgA1 by the monoclonal antibody KM55 contributes to predicting patients with IgA nephropathy with high risk of long-term progression. Nefrologia (Engl Ed) 41:311–320. https://doi.org/10.1016/j.nefro.2020.12.011
Yuzawa Y, Yamamoto R, Takahashi K, Katafuchi R, Tomita M, Fujigaki Y, Kitamura H, Goto M, Yasuda T, Sato M, Urushihara M, Kondo S, Kagami S, Yasuda Y, Komatsu H, Takahara M, Harabuchi Y, Kimura K, Matsuo S (2016) Evidence-based clinical practice guidelines for IgA nephropathy 2014. Clin Exp Nephrol 20:511–535. https://doi.org/10.1007/s10157-015-1223-y
Thuluvath PJ, Ninkovic M, Calam J, Anderson M (1994) Mesalazine induced interstitial nephritis Gut 35:1493–1496. https://doi.org/10.1136/gut.35.10.1493
Feehally J, Khosravi M (2015) Effects of acute and chronic hypohydration on kidney health and function. Nutr Rev 73(Suppl 2):110–119. https://doi.org/10.1093/nutrit/nuv046
Wijkstrom J, Gonzalez-Quiroz M, Hernandez M, Trujillo Z, Hultenby K, Ring A, Soderberg M, Aragon A, Elinder CG, Wernerson A (2017) Renal morphology, clinical findings, and progression rate in Mesoamerican nephropathy. Am J Kidney Dis 69:626–636. https://doi.org/10.1053/j.ajkd.2016.10.036
Momoki K, Kataoka H, Moriyama T, Mochizuki T, Nitta K (2017) Hyperuricemia as a predictive marker for progression of nephrosclerosis: clinical assessment of prognostic factors in biopsy-proven arterial/arteriolar nephrosclerosis. J Atheroscler Thromb 24:630–642. https://doi.org/10.5551/jat.37523
Adiga A, Goldfarb DS (2020) The association of mesalamine with kidney disease. Adv Chronic Kidney Dis 27:72–76. https://doi.org/10.1053/j.ackd.2019.09.002
Heap GA, So K, Weedon M, Edney N, Bewshea C, Singh A, Annese V, Beckly J, Buurman D, Chaudhary R, Cole AT, Cooper SC, Creed T, Cummings F, de Boer NK, D’Inca R, D’Souza R, Daneshmend TK, Delaney M, Dhar A, Direkze N, Dunckley P, Gaya DR, Gearry R, Gore S, Halfvarson J, Hart A, Hawkey CJ, Hoentjen F, Iqbal T, Irving P, Lal S, Lawrance I, Lees CW, Lockett M, Mann S, Mansfield J, Mowat C, Mulgrew CJ, Muller F, Murray C, Oram R, Orchard T, Parkes M, Phillips R, Pollok R, Radford-Smith G, Sebastian S, Sen S, Shirazi T, Silverberg M, Solomon L, Sturniolo GC, Thomas M, Tremelling M, Tsianos EV, Watts D, Weaver S, Weersma RK, Wesley E, Holden A, Ahmad T (2016) Clinical features and HLA association of 5-aminosalicylate (5-ASA)-induced nephrotoxicity in inflammatory bowel disease. J Crohns Colitis 10:149–158. https://doi.org/10.1093/ecco-jcc/jjv219
Oikonomou KA, Kapsoritakis AN, Stefanidis I, Potamianos SP (2011) Drug-induced nephrotoxicity in inflammatory bowel disease. Nephron Clin Pract 119:c89–94; discussion c96. https://doi.org/10.1159/000326682
Waters AM, Zachos M, Herzenberg AM, Harvey E, Rosenblum ND (2008) Tubulointerstitial nephritis as an extraintestinal manifestation of Crohn’s disease. Nat Clin Pract Nephrol 4:693–697. https://doi.org/10.1038/ncpneph0955
Izzedine H, Simon J, Piette AM, Lucsko M, Baumelou A, Charitanski D, Kernaonet E, Baglin AC, Deray G, Beaufils H (2002) Primary chronic interstitial nephritis in Crohn’s disease. Gastroenterology 123:1436–1440. https://doi.org/10.1053/gast.2002.36613
Li G, Wu W, Zhang X, Huang Y, Wen Y, Li X, Gao R (2018) Serum levels of tumor necrosis factor alpha in patients with IgA nephropathy are closely associated with disease severity. BMC Nephrol 19:326. https://doi.org/10.1186/s12882-018-1069-0
Salvador P, Macias-Ceja DC, Gisbert-Ferrandiz L, Hernandez C, Bernardo D, Alos R, Navarro-Vicente F, Esplugues JV, Ortiz-Masia D, Barrachina MD, Calatayud S (2018) CD16+ macrophages mediate fibrosis in inflammatory bowel disease. J Crohns Colitis 12:589–599. https://doi.org/10.1093/ecco-jcc/jjx185
Sieber G, Herrmann F, Zeitz M, Teichmann H, Ruhl H (1984) Abnormalities of B-cell activation and immunoregulation in patients with Crohn’s disease. Gut 25:1255–1261. https://doi.org/10.1136/gut.25.11.1255
Uzzan M, Colombel JF, Cerutti A, Treton X, Mehandru S (2016) B cell-activating factor (BAFF)-targeted B cell therapies in inflammatory bowel diseases. Dig Dis Sci 61:3407–3424. https://doi.org/10.1007/s10620-016-4317-9
Heller F, Lindenmeyer MT, Cohen CD, Brandt U, Draganovici D, Fischereder M, Kretzler M, Anders HJ, Sitter T, Mosberger I, Kerjaschki D, Regele H, Schlondorff D, Segerer S (2007) The contribution of B cells to renal interstitial inflammation. Am J Pathol 170:457–468. https://doi.org/10.2353/ajpath.2007.060554
Xavier S, Sahu RK, Landes SG, Yu J, Taylor RP, Ayyadevara S, Megyesi J, Stallcup WB, Duffield JS, Reis ES, Lambris JD, Portilla D (2017) Pericytes and immune cells contribute to complement activation in tubulointerstitial fibrosis. Am J Physiol Renal Physiol 312:F516–F532. https://doi.org/10.1152/ajprenal.00604.2016
Liu Y, Wang K, Liang X, Li Y, Zhang Y, Zhang C, Wei H, Luo R, Ge S, Xu G (2018) Complement C3 produced by macrophages promotes renal fibrosis via IL-17A secretion. Front Immunol 9:2385. https://doi.org/10.3389/fimmu.2018.02385
Halstensen TS, Mollnes TE, Garred P, Fausa O, Brandtzaeg P (1992) Surface epithelium related activation of complement differs in Crohn’s disease and ulcerative colitis. Gut 33:902–908. https://doi.org/10.1136/gut.33.7.902
Sugihara T, Kobori A, Imaeda H, Tsujikawa T, Amagase K, Takeuchi K, Fujiyama Y, Andoh A (2010) The increased mucosal mRNA expressions of complement C3 and interleukin-17 in inflammatory bowel disease. Clin Exp Immunol 160:386–393. https://doi.org/10.1111/j.1365-2249.2010.04093.x
Yang P, Chen X, Zeng L, Hao H, Xu G (2017) The response of the Oxford classification to steroid in IgA nephropathy: a systematic review and meta-analysis. Oncotarget 8:59748–59756. https://doi.org/10.18632/oncotarget.19574
Acknowledgements
We greatly appreciate the excellent technical assistance for immunohistochemistry provided by Ms. Tomoko Suzuki, Division of Nephrology, Department of Medicine, Showa University School of Medicine and Mr. Toshiki Tadenuma, Clinical Laboratory, Tokyo Yamate Medical Center. This study was presented in the Annual Meeting of ASN 2020.
Author information
Authors and Affiliations
Contributions
M.A., K.H., and H.Y. conceived and designed the study. M.A. created figures. M.A., K.H., M.K., and K.A. reviewed the histology. M.A., K.H., K.S., H.Y., and M.S. collected data. M.A. and K.H. analyzed and interpreted data. M.A., K.H., M.K., K.A., Y.W., and M.I. drafted and finalized the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Akiyama, M., Shimomura, K., Yoshimoto, H. et al. Crohn’s disease may promote inflammation in IgA nephropathy: a case–control study of patients undergoing kidney biopsy. Virchows Arch 481, 553–563 (2022). https://doi.org/10.1007/s00428-022-03373-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-022-03373-w