
Mutated KIT Tyrosine Kinase as a Novel Molecular Target in Acute Myeloid Leukemia

 , , , , ,
, , , , ,
Abstract
:1. Introduction
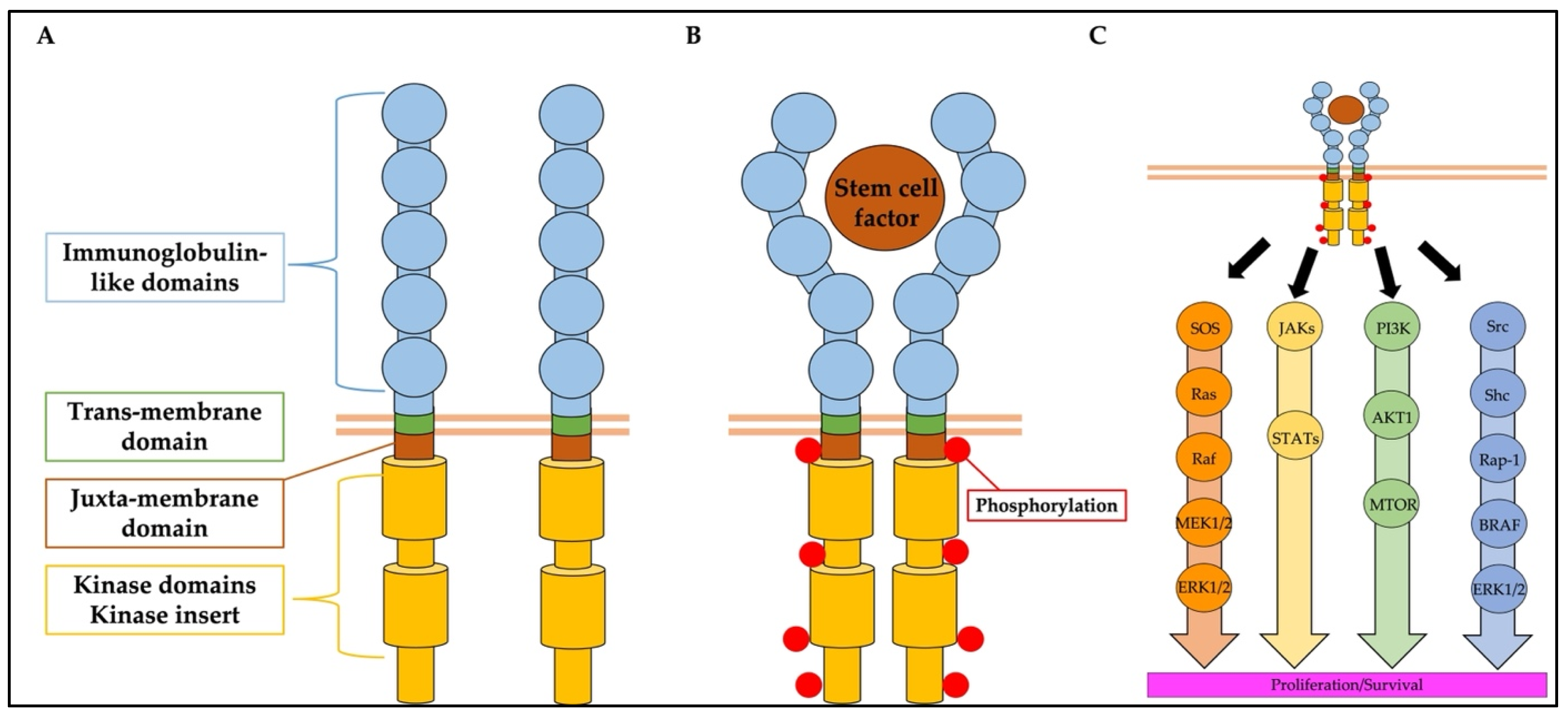
2. Structure, Function, and Mutation of KIT
2.1. Structure and Function of KIT
2.2. Mutations of KIT in Cancer
3. Prognosis of AML with KIT Mutations Treated with Conventional Chemotherapy
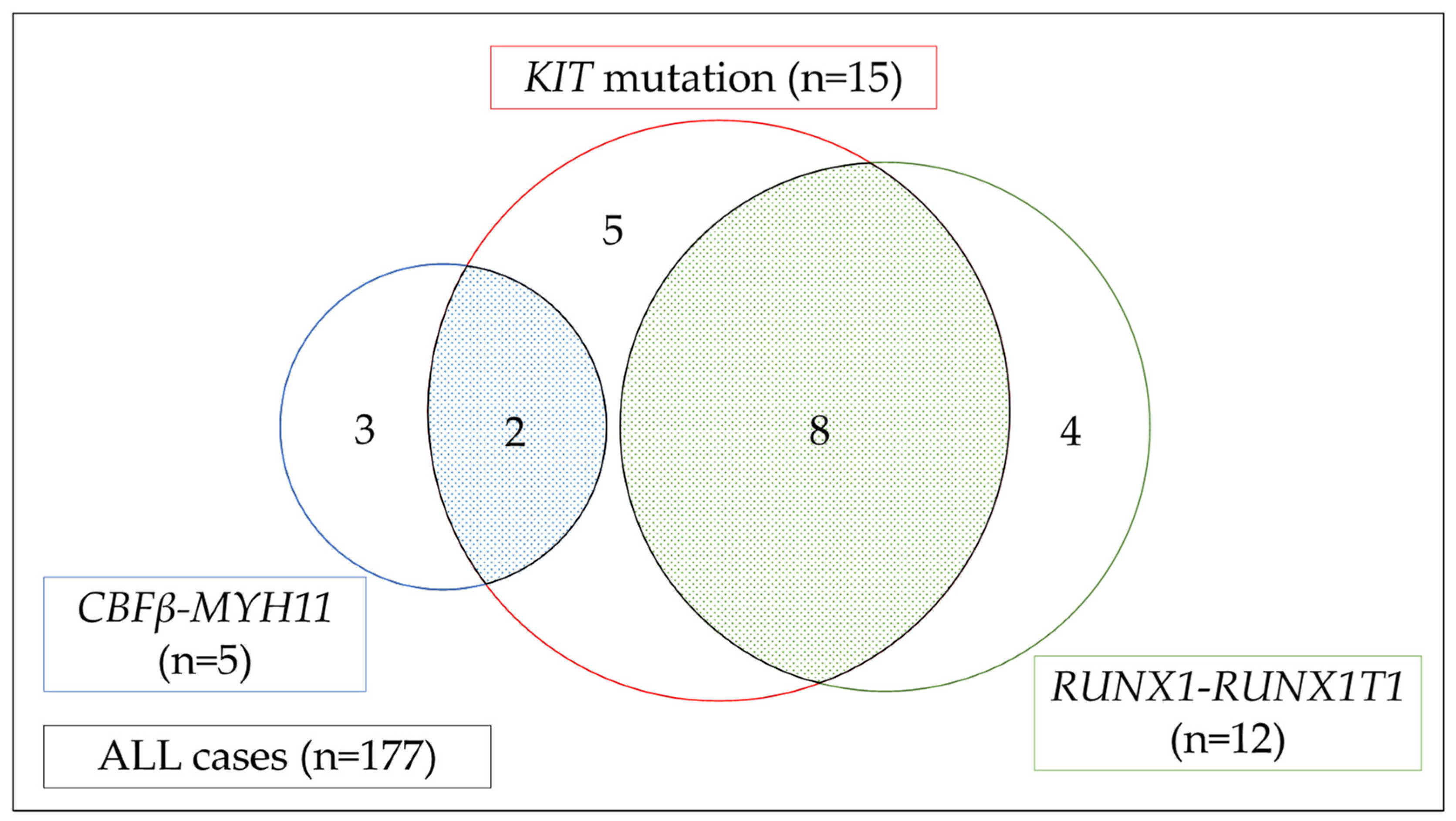
4. KIT Mutation in Unfit and Relapsed/Refractory AML: Results from the HM-SCREEN-Japan-01 Study
4.1. Frequency of KIT Mutation in Unfit and R/R AML
4.2. Landscape of Gene Mutations in the KIT Mutation Cohort
4.3. Clinical Impact of KIT Mutation in Unfit and R/R AML
5. Possible Role for Kinase Inhibitors in the Treatment of AML with KIT Mutation
6. HSP90 Inhibitors for the Treatment of AML with KIT Mutation
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miettinen, M.; Lasota, J. KIT (CD117): A review on expression in normal and neoplastic tissues, and mutations and their clinicopathologic correlation. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 205–220. [Google Scholar] [CrossRef]
- Shomali, W.; Gotlib, J. The new tool “KIT” in advanced systemic mastocytosis. Hematol. Am. Soc. Hematol. Educ. Program. 2018, 2018, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Lennartsson, J.; Rönnstrand, L. Stem cell factor receptor/c-Kit: From basic science to clinical implications. Physiol. Rev. 2012, 92, 1619–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, B.P.; Heinrich, M.C.; Corless, C.L. Gastrointestinal stromal tumour. Lancet 2007, 369, 1731–1741. [Google Scholar] [CrossRef]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Castañeda, L.D.; Nova, J.A.; Tovar-Parra, J.D. Frequency of mutations in BRAF, NRAS, and KIT in different populations and histological subtypes of melanoma: A systemic review. Melanoma Res. 2020, 30, 62–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauth, M.T.; Eder, C.; Alpermann, T.; Bacher, U.; Nadarajah, N.; Kern, W.; Haferlach, C.; Haferlach, T.; Schnittger, S. High number of additional genetic lesions in acute myeloid leukemia with t(8;21)/RUNX1-RUNX1T1: Frequency and impact on clinical outcome. Leukemia 2014, 28, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.Z.; Zhu, H.H.; Jiang, Q.; Jiang, H.; Zhang, L.P.; Xu, L.P.; Wang, Y.; Liu, Y.R.; Lai, Y.Y.; Shi, H.X.; et al. Prevalence and prognostic significance of c-KIT mutations in core binding factor acute myeloid leukemia: A comprehensive large-scale study from a single Chinese center. Leuk. Res. 2014, 38, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Hills, R.K.; Lamb, K.; Evans, C.; Tinsley, S.; Sellar, R.; O’Brien, M.; Yin, J.L.; Burnett, A.K.; Linch, D.C.; et al. The importance of relative mutant level for evaluating impact on outcome of KIT, FLT3 and CBL mutations in core-binding factor acute myeloid leukemia. Leukemia 2013, 27, 1891–1901. [Google Scholar] [CrossRef]
- Paschka, P.; Du, J.; Schlenk, R.F.; Gaidzik, V.I.; Bullinger, L.; Corbacioglu, A.; Späth, D.; Kayser, S.; Schlegelberger, B.; Krauter, J.; et al. Secondary genetic lesions in acute myeloid leukemia with inv(16) or t(16;16): A study of the German-Austrian AML Study Group (AMLSG). Blood 2013, 121, 170–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Ahn, H.K.; Jung, C.W.; Moon, J.H.; Park, C.H.; Lee, K.O.; Kim, S.H.; Kim, Y.K.; Kim, H.J.; Sohn, S.K.; et al. KIT D816 mutation associates with adverse outcomes in core binding factor acute myeloid leukemia, especially in the subgroup with RUNX1/RUNX1T1 rearrangement. Ann. Hematol. 2013, 92, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [PubMed] [Green Version]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [Green Version]
- Burd, A.; Levine, R.L.; Ruppert, A.S.; Mims, A.S.; Borate, U.; Stein, E.M.; Patel, P.; Baer, M.R.; Stock, W.; Deininger, M.; et al. Precision medicine treatment in acute myeloid leukemia using prospective genomic profiling: Feasibility and preliminary efficacy of the Beat AML Master Trial. Nat. Med. 2020, 26, 1852–1858. [Google Scholar] [CrossRef]
- Yee, N.S.; Hsiau, C.W.; Serve, H.; Vosseller, K.; Besmer, P. Mechanism of down-regulation of c-kit receptor. Roles of receptor tyrosine kinase, phosphatidylinositol 3′-kinase, and protein kinase C. J. Biol. Chem. 1994, 269, 31991–31998. [Google Scholar] [CrossRef]
- Yuzawa, S.; Opatowsky, Y.; Zhang, Z.; Mandiyan, V.; Lax, I.; Schlessinger, J. Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell 2007, 130, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Sattler, M.; Salgia, R. Targeting c-Kit mutations: Basic science to novel therapies. Leuk. Res. 2004, 28 (Suppl. 1), S11–S20. [Google Scholar] [CrossRef]
- Pathania, S.; Pentikäinen, O.T.; Singh, P.K. A holistic view on c-Kit in cancer: Structure, signaling, pathophysiology and its inhibitors. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188631. [Google Scholar] [CrossRef]
- Rottapel, R.; Reedijk, M.; Williams, D.E.; Lyman, S.D.; Anderson, D.M.; Pawson, T.; Bernstein, A. The Steel/W transduction pathway: Kit autophosphorylation and its association with a unique subset of cytoplasmic signaling proteins is induced by the Steel factor. Mol. Cell Biol. 1991, 11, 3043–3051. [Google Scholar]
- Serve, H.; Hsu, Y.C.; Besmer, P. Tyrosine residue 719 of the c-kit receptor is essential for binding of the P85 subunit of phosphatidylinositol (PI) 3-kinase and for c-kit-associated PI 3-kinase activity in COS-1 cells. J. Biol. Chem. 1994, 269, 6026–6030. [Google Scholar] [CrossRef]
- Weiler, S.R.; Mou, S.; DeBerry, C.S.; Keller, J.R.; Ruscetti, F.W.; Ferris, D.K.; Longo, D.L.; Linnekin, D. JAK2 is associated with the c-kit proto-oncogene product and is phosphorylated in response to stem cell factor. Blood 1996, 87, 3688–3693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotoh, A.; Takahira, H.; Mantel, C.; Litz-Jackson, S.; Boswell, H.S.; Broxmeyer, H.E. Steel factor induces serine phosphorylation of Stat3 in human growth factor-dependent myeloid cell lines. Blood 1996, 88, 138–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Zoller, K.; Masuko, M.; Rojnuckarin, P.; Yang, X.O.; Parganas, E.; Kaushansky, K.; Ihle, J.N.; Papayannopoulou, T.; Willerford, D.M.; et al. JAK2, complemented by a second signal from c-kit or flt-3, triggers extensive self-renewal of primary multipotential hemopoietic cells. Embo J. 2002, 21, 2159–2167. [Google Scholar] [CrossRef] [Green Version]
- Wandzioch, E.; Edling, C.E.; Palmer, R.H.; Carlsson, L.; Hallberg, B. Activation of the MAP kinase pathway by c-Kit is PI-3 kinase dependent in hematopoietic progenitor/stem cell lines. Blood 2004, 104, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Lennartsson, J.; Blume-Jensen, P.; Hermanson, M.; Pontén, E.; Carlberg, M.; Rönnstrand, L. Phosphorylation of Shc by Src family kinases is necessary for stem cell factor receptor/c-kit mediated activation of the Ras/MAP kinase pathway and c-fos induction. Oncogene 1999, 18, 5546–5553. [Google Scholar] [CrossRef] [Green Version]
- Thömmes, K.; Lennartsson, J.; Carlberg, M.; Rönnstrand, L. Identification of Tyr-703 and Tyr-936 as the primary association sites for Grb2 and Grb7 in the c-Kit/stem cell factor receptor. Biochem. J. 1999, 341 Pt 1, 211–216. [Google Scholar] [CrossRef]
- Voytyuk, O.; Lennartsson, J.; Mogi, A.; Caruana, G.; Courtneidge, S.; Ashman, L.K.; Rönnstrand, L. Src family kinases are involved in the differential signaling from two splice forms of c-Kit. J. Biol. Chem. 2003, 278, 9159–9166. [Google Scholar] [CrossRef] [Green Version]
- Edling, C.E.; Hallberg, B. c-Kit—A hematopoietic cell essential receptor tyrosine kinase. Int. J. Biochem. Cell Biol. 2007, 39, 1995–1998. [Google Scholar] [CrossRef]
- Ogawa, M.; Matsuzaki, Y.; Nishikawa, S.; Hayashi, S.; Kunisada, T.; Sudo, T.; Kina, T.; Nakauchi, H.; Nishikawa, S. Expression and function of c-kit in hemopoietic progenitor cells. J. Exp. Med. 1991, 174, 63–71. [Google Scholar] [CrossRef]
- Bowie, M.B.; Kent, D.G.; Copley, M.R.; Eaves, C.J. Steel factor responsiveness regulates the high self-renewal phenotype of fetal hematopoietic stem cells. Blood 2007, 109, 5043–5048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.Y.; Hu, W.; Naramura, M.; Park, C.Y. High c-Kit expression identifies hematopoietic stem cells with impaired self-renewal and megakaryocytic bias. J. Exp. Med. 2014, 211, 217–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruse, G.; Metcalfe, D.D.; Olivera, A. Functional deregulation of KIT: Link to mast cell proliferative diseases and other neoplasms. Immunol. Allergy Clin. N. Am. 2014, 34, 219–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, M.C.; Blanke, C.D.; Druker, B.J.; Corless, C.L. Inhibition of KIT tyrosine kinase activity: A novel molecular approach to the treatment of KIT-positive malignancies. J. Clin. Oncol. 2002, 20, 1692–1703. [Google Scholar] [CrossRef]
- Montone, K.T.; van Belle, P.; Elenitsas, R.; Elder, D.E. Proto-oncogene c-kit expression in malignant melanoma: Protein loss with tumor progression. Mod. Pathol. 1997, 10, 939–944. [Google Scholar] [PubMed]
- Liang, J.; Wu, Y.L.; Chen, B.J.; Zhang, W.; Tanaka, Y.; Sugiyama, H. The C-kit receptor-mediated signal transduction and tumor-related diseases. Int. J. Biol. Sci. 2013, 9, 435–443. [Google Scholar] [CrossRef]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef]
- Hongyo, T.; Li, T.; Syaifudin, M.; Baskar, R.; Ikeda, H.; Kanakura, Y.; Aozasa, K.; Nomura, T. Specific c-kit mutations in sinonasal natural killer/T-cell lymphoma in China and Japan. Cancer Res. 2000, 60, 2345–2347. [Google Scholar]
- Nagata, H.; Worobec, A.S.; Oh, C.K.; Chowdhury, B.A.; Tannenbaum, S.; Suzuki, Y.; Metcalfe, D.D. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc. Natl. Acad. Sci. USA 1995, 92, 10560–10564. [Google Scholar] [CrossRef] [Green Version]
- Longley, B.J.; Tyrrell, L.; Lu, S.Z.; Ma, Y.S.; Langley, K.; Ding, T.G.; Duffy, T.; Jacobs, P.; Tang, L.H.; Modlin, I. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: Establishment of clonality in a human mast cell neoplasm. Nat. Genet. 1996, 12, 312–314. [Google Scholar] [CrossRef]
- Tian, Q.; Frierson, H.F., Jr.; Krystal, G.W.; Moskaluk, C.A. Activating c-kit gene mutations in human germ cell tumors. Am. J. Pathol. 1999, 154, 1643–1647. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.P.; Gönen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Gao, L.; Chen, J.; Hu, S. Influence of KIT mutations on prognosis of pediatric patients with core-binding factor acute myeloid leukemia: A systematic review and meta-analysis. Transl. Pediatr. 2020, 9, 726–733. [Google Scholar] [CrossRef]
- Paschka, P.; Döhner, K. Core-binding factor acute myeloid leukemia: Can we improve on HiDAC consolidation? Hematol. Am. Soc. Hematol. Educ. Program. 2013, 2013, 209–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, Y. Molecular pathogenesis and treatment of core binding factor-acute myeloid leukemia. Jpn. J. Clin. Hematol. 2018, 59, 1997–2006. [Google Scholar]
- Kim, S.Y.; Kang, J.J.; Lee, H.H.; Kang, J.J.; Kim, B.; Kim, C.G.; Park, T.K.; Kang, H. Mechanism of activation of human c-KIT kinase by internal tandem duplications of the juxtamembrane domain and point mutations at aspartic acid 816. Biochem. Biophys. Res. Commun. 2011, 410, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Berenstein, R. Class III Receptor Tyrosine Kinases in Acute Leukemia—Biological Functions and Modern Laboratory Analysis. Biomark. Insights 2015, 10 (Suppl. 3), 1–14. [Google Scholar] [CrossRef] [Green Version]
- Schnittger, S.; Kohl, T.M.; Haferlach, T.; Kern, W.; Hiddemann, W.; Spiekermann, K.; Schoch, C. KIT-D816 mutations in AML1-ETO-positive AML are associated with impaired event-free and overall survival. Blood 2006, 107, 1791–1799. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, Y.; Kawashima, N.; Atsuta, Y.; Sugiura, I.; Sawa, M.; Dobashi, N.; Yokoyama, H.; Doki, N.; Tomita, A.; Kiguchi, T.; et al. Prospective evaluation of prognostic impact of KIT mutations on acute myeloid leukemia with RUNX1-RUNX1T1 and CBFB-MYH11. Blood Adv. 2020, 4, 66–75. [Google Scholar] [CrossRef]
- Wichmann, C.; Quagliano-Lo Coco, I.; Yildiz, Ö.; Chen-Wichmann, L.; Weber, H.; Syzonenko, T.; Döring, C.; Brendel, C.; Ponnusamy, K.; Kinner, A.; et al. Activating c-KIT mutations confer oncogenic cooperativity and rescue RUNX1/ETO-induced DNA damage and apoptosis in human primary CD34+ hematopoietic progenitors. Leukemia 2015, 29, 279–289. [Google Scholar] [CrossRef]
- Omori, I.; Yamaguchi, H.; Miyake, K.; Miyake, N.; Kitano, T.; Inokuchi, K. D816V mutation in the KIT gene activation loop has greater cell-proliferative and anti-apoptotic ability than N822K mutation in core-binding factor acute myeloid leukemia. Exp. Hematol. 2017, 52, 56–64.e4. [Google Scholar] [CrossRef] [PubMed]
- Tarlock, K.; Alonzo, T.A.; Wang, Y.C.; Gerbing, R.B.; Ries, R.; Loken, M.R.; Pardo, L.; Hylkema, T.; Joaquin, J.; Sarukkai, L.; et al. Functional Properties of KIT Mutations Are Associated with Differential Clinical Outcomes and Response to Targeted Therapeutics in CBF Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 5038–5048. [Google Scholar] [CrossRef] [PubMed]
- Hosono, N.; Yamauchi, T.; Chi, S.; Fukushima, K.; Shibayama, H.; Katagiri, S.; Gotoh, A.; Eguchi, M.; Morishita, T.; Ogasawara, R.; et al. Hematologic Malignancies (HM)-Screen-Japan 01: A Mutation Profiling Multicenter Study on Patients with Acute Myeloid Leukemia. Blood 2021, 138 (Suppl. 1), 4457. [Google Scholar] [CrossRef]
- Miyamoto, K.; Minami, Y. Precision medicine and novel molecular target therapies in acute myeloid leukemia: The background of hematologic malignancies (HM)-SCREEN-Japan 01. Int. J. Clin. Oncol. 2019, 24, 893–898. [Google Scholar] [CrossRef] [Green Version]
- Katagiri, S.; Akahane, D.; Gotoh, A.; Chi, S.; Fukushima, K.; Shibayama, H.; Hosono, N.; Yamauchi, T.; Eguchi, M.; Morishita, T.; et al. Genomic Analysis Focusing on RUNX1-RUNX1T1 in Japanese Patients with AML: HM-Screen-Japan 01. Blood 2021, 138 (Suppl. 1), 4464. [Google Scholar] [CrossRef]
- Carter, J.L.; Hege, K.; Yang, J.; Kalpage, H.A.; Su, Y.; Edwards, H.; Hüttemann, M.; Taub, J.W.; Ge, Y. Targeting multiple signaling pathways: The new approach to acute myeloid leukemia therapy. Signal Transduct. Target Ther. 2020, 5, 288. [Google Scholar] [CrossRef]
- Klug, L.R.; Corless, C.L.; Heinrich, M.C. Inhibition of KIT Tyrosine Kinase Activity: Two Decades After the First Approval. J. Clin. Oncol. 2021, 39, 1674–1686. [Google Scholar] [CrossRef]
- Advani, A.S.; Tiu, R.; Saunthararajah, Y.; Maciejewski, J.; Copelan, E.A.; Sobecks, R.; Sekeres, M.A.; Bates, J.; Rush, M.L.; Tripp, B.; et al. A Phase 1 study of imatinib mesylate in combination with cytarabine and daunorubicin for c-kit positive relapsed acute myeloid leukemia. Leuk. Res. 2010, 34, 1622–1626. [Google Scholar] [CrossRef]
- Brandwein, J.M.; Hedley, D.W.; Chow, S.; Schimmer, A.D.; Yee, K.W.; Schuh, A.C.; Gupta, V.; Xu, W.; Kamel-Reid, S.; Minden, M.D. A phase I/II study of imatinib plus reinduction therapy for c-kit-positive relapsed/refractory acute myeloid leukemia: Inhibition of Akt activation correlates with complete response. Leukemia 2011, 25, 945–952. [Google Scholar] [CrossRef]
- Heidel, F.; Cortes, J.; Rücker, F.G.; Aulitzky, W.; Letvak, L.; Kindler, T.; Huber, C.; Döhner, H.; Kantarjian, H.; Fischer, T. Results of a multicenter phase II trial for older patients with c-Kit-positive acute myeloid leukemia (AML) and high-risk myelodysplastic syndrome (HR-MDS) using low-dose Ara-C and Imatinib. Cancer 2007, 109, 907–914. [Google Scholar] [CrossRef]
- Advani, A.S.; Tse, W.; Li, H.; Jia, X.; Elson, P.; Cooper, B.; Ali-Osman, F.; Park, J.; Rao, A.V.; Rizzieri, D.A.; et al. A Phase II Trial of Imatinib Mesylate as Maintenance Therapy for Patients with Newly Diagnosed C-kit-positive Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2021, 21, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S. Expanding dasatinib beyond KIT in acute myeloid leukemia. Haematologica 2020, 105, 2708–2710. [Google Scholar] [CrossRef] [PubMed]
- Malani, D.; Yadav, B.; Kumar, A.; Potdar, S.; Kontro, M.; Kankainen, M.; Javarappa, K.K.; Porkka, K.; Wolf, M.; Aittokallio, T.; et al. KIT pathway upregulation predicts dasatinib efficacy in acute myeloid leukemia. Leukemia 2020, 34, 2780–2784. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Geyer, S.; Laumann, K.; Zhao, W.; Bucci, D.; Uy, G.L.; Blum, W.; Eisfeld, A.K.; Pardee, T.S.; Wang, E.S.; et al. Combination of dasatinib with chemotherapy in previously untreated core binding factor acute myeloid leukemia: CALGB 10801. Blood Adv. 2020, 4, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Paschka, P.; Schlenk, R.F.; Weber, D.; Benner, A.; Bullinger, L.; Heuser, M.; Gaidzik, V.I.; Thol, F.; Agrawal, M.; Teleanu, V.; et al. Adding dasatinib to intensive treatment in core-binding factor acute myeloid leukemia-results of the AMLSG 11-08 trial. Leukemia 2018, 32, 1621–1630. [Google Scholar] [CrossRef]
- Weisberg, E.; Boulton, C.; Kelly, L.M.; Manley, P.; Fabbro, D.; Meyer, T.; Gilliland, D.G.; Griffin, J.D. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell 2002, 1, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Gotlib, J.; Berubé, C.; Growney, J.D.; Chen, C.C.; George, T.I.; Williams, C.; Kajiguchi, T.; Ruan, J.; Lilleberg, S.L.; Durocher, J.A.; et al. Activity of the tyrosine kinase inhibitor PKC412 in a patient with mast cell leukemia with the D816V KIT mutation. Blood 2005, 106, 2865–2870. [Google Scholar] [CrossRef] [Green Version]
- Gleixner, K.V.; Mayerhofer, M.; Aichberger, K.J.; Derdak, S.; Sonneck, K.; Böhm, A.; Gruze, A.; Samorapoompichit, P.; Manley, P.W.; Fabbro, D.; et al. PKC412 inhibits in vitro growth of neoplastic human mast cells expressing the D816V-mutated variant of KIT: Comparison with AMN107, imatinib, and cladribine (2CdA) and evaluation of cooperative drug effects. Blood 2006, 107, 752–759. [Google Scholar] [CrossRef] [Green Version]
- Miyata, Y.; Nakamoto, H.; Neckers, L. The therapeutic target Hsp90 and cancer hallmarks. Curr. Pharm. Des. 2013, 19, 347–365. [Google Scholar] [CrossRef]
- Banerji, U. Heat shock protein 90 as a drug target: Some like it hot. Clin. Cancer Res. 2009, 15, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Solárová, Z.; Mojžiš, J.; Solár, P. Hsp90 inhibitor as a sensitizer of cancer cells to different therapies (review). Int. J. Oncol. 2015, 46, 907–926. [Google Scholar] [PubMed] [Green Version]
- Ohkubo, S.; Kodama, Y.; Muraoka, H.; Hitotsumachi, H.; Yoshimura, C.; Kitade, M.; Hashimoto, A.; Ito, K.; Gomori, A.; Takahashi, K.; et al. TAS-116, a highly selective inhibitor of heat shock protein 90α and β, demonstrates potent antitumor activity and minimal ocular toxicity in preclinical models. Mol. Cancer Ther. 2015, 14, 14–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prodromou, C.; Roe, S.M.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 1997, 90, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Ikebe, E.; Shimosaki, S.; Hasegawa, H.; Iha, H.; Tsukamoto, Y.; Wang, Y.; Sasaki, D.; Imaizumi, Y.; Miyazaki, Y.; Yanagihara, K.; et al. TAS-116 (pimitespib), a heat shock protein 90 inhibitor, shows efficacy in preclinical models of adult T-cell leukemia. Cancer Sci. 2022, 113, 684–696. [Google Scholar] [CrossRef]
- Walsby, E.J.; Lazenby, M.; Pepper, C.J.; Knapper, S.; Burnett, A.K. The HSP90 inhibitor NVP-AUY922-AG inhibits the PI3K and IKK signalling pathways and synergizes with cytarabine in acute myeloid leukaemia cells. Br. J. Haematol. 2013, 161, 57–67. [Google Scholar] [CrossRef]
- Al Shaer, L.; Walsby, E.; Gilkes, A.; Tonks, A.; Walsh, V.; Mills, K.; Burnett, A.; Rowntree, C. Heat shock protein 90 inhibition is cytotoxic to primary AML cells expressing mutant FLT3 and results in altered downstream signalling. Br. J. Haematol. 2008, 141, 483–493. [Google Scholar] [CrossRef]
- Minami, Y.; Kiyoi, H.; Yamamoto, Y.; Yamamoto, K.; Ueda, R.; Saito, H.; Naoe, T. Selective apoptosis of tandemly duplicated FLT3-transformed leukemia cells by Hsp90 inhibitors. Leukemia 2002, 16, 1535–1540. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Wang, J.; Jin, J.; Qian, W.; Qian, J.; Cheng, Y.; Wang, L. Heat shock protein 90 inhibition results in altered downstream signaling of mutant KIT and exerts synergistic effects on Kasumi-1 cells when combining with histone deacetylase inhibitor. Leuk. Res. 2011, 35, 1212–1218. [Google Scholar] [CrossRef]
- Tsujimura, A.; Kiyoi, H.; Shiotsu, Y.; Ishikawa, Y.; Mori, Y.; Ishida, H.; Toki, T.; Ito, E.; Naoe, T. Selective KIT inhibitor KI-328 and HSP90 inhibitor show different potency against the type of KIT mutations recurrently identified in acute myeloid leukemia. Int. J. Hematol. 2010, 92, 624–633. [Google Scholar] [CrossRef]
- Workman, P.; Burrows, F.; Neckers, L.; Rosen, N. Drugging the cancer chaperone HSP90: Combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann. N. Y. Acad. Sci. 2007, 1113, 202–216. [Google Scholar] [CrossRef]
- Saito, Y.; Takahashi, T.; Obata, Y.; Nishida, T.; Ohkubo, S.; Nakagawa, F.; Serada, S.; Fujimoto, M.; Ohkawara, T.; Nishigaki, T.; et al. TAS-116 inhibits oncogenic KIT signalling on the Golgi in both imatinib-naïve and imatinib-resistant gastrointestinal stromal tumours. Br. J. Cancer 2020, 122, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Honma, Y.; Kurokawa, Y.; Sawaki, A.; Naito, Y.; Iwagami, S.; Baba, H.; Komatsu, Y.; Nishida, T.; Doi, T. Randomized, double-blind, placebo (PL)-controlled, phase III trial of pimitespib (TAS-116), an oral inhibitor of heat shock protein 90 (HSP90), in patients (pts) with advanced gastrointestinal stromal tumor (GIST) refractory to imatinib (IM), sunitinib (SU) and regorafenib (REG). J. Clin. Oncol. 2021, 39 (Suppl. 15), 11524. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Exon | Disease | Description |
|---|---|---|---|
| Immunoglobulin-like domain | 8 | AML | T417, Y418, D419 |
| 9 | GIST | A502 | |
| Mastocytosis | K5091 | ||
| Trans-membrane domain | 10 | AML | V530I |
| Mastocytosis | F522C, A533D | ||
| Juxta-membrane domain | 11 | AML | V560, V559, ITD |
| GIST | CD117, V559A, V559D, W557R, V560G | ||
| Melanoma | L576P | ||
| Mastocytosis | V560G | ||
| 13 | AML | K642E | |
| Melanoma | K642E | ||
| 14 | GIST | K704, N705 | |
| Kinase insert | 15 | GIST | S715 |
| Kinase domain | 16 | AML | 1748T, L773S |
| 17 | AML | D816V, D816Y, D816F, D816H, N822, V8251 | |
| Germ cell tumor | D816H, D816V | ||
| Mastocytosis | D816V, D816Y, D816H, D820G | ||
| ENKL | V825A, D816N |
| Exon | Description | Functional Impact |
|---|---|---|
| 8 | T417, Y418, D419 | Hyper-reactivity to stem cell factor |
| 10–11 | V530, V540, W557, V559, L576, ITD | Spontaneous dimer formation |
| 17 | D816, D820, N822, Y823, V825 | Auto activation |
| Author, Year | Disease Status | Frequency of KIT Mutations | ||
|---|---|---|---|---|
| CBF Leukemia | RUNX1-RUNX1T1 | CBFβ-MYH11 | ||
| Qin 2014 | Newly diagnosed | 37% (128/351) | 39% (99/253) | 30% (29/98) |
| Allen 2013 | Newly diagnosed | 28% (100/354) | 23% (46/199) | 35% (54/155) |
| Kim 2013 | Newly diagnosed | 26% (32/121) | 27% (22/82) | 35% (54/155) |
| Ishikawa 2019 | Newly diagnosed | 34% (63/199) | 32% (42/132) | 31% (21/67) |
| HM-SCREEN01 | R/R or Unfit | 59% (10/17) | 67% (8/12) | 40% (2/5) |
| KIT Mutation | ||||||
|---|---|---|---|---|---|---|
| ID | Category | Description | SNV | VAF | Other Mutations | Chromosomal Karyotype |
| 13 | Unfit | D816V | 2447A > T | 0.372 | FLT3, KRAS, CEBPA, TP53 | Complex karyotype |
| 50 | Unfit | D816V | 2447A > T | 0.123 | RAD21, SPEN | t(8;21)(q22;q22.1) |
| 56 | Unfit | D816F | 2446_2447GA > TT | 0.37 | TP53, CDKN2A, CDKN2B | Complex karyotype |
| 149 | Unfit | D816V | 2447A > T | 0.344 | ASXL1, DNMT3A, SETBP1, FANCD2, CASP8 | 46, XY, t(3;3)(p25:q13) |
| 158 | Unfit | T417_D419 > Y | 1249_1255ACTTACG > T | 0.078 | FLT3, NRAS | inv(16)/t(16;16) |
| 160 | Unfit | D816V | 2447A > T | 0.146 | CSF3R, JAK1 | t(8;21)(q22;q22.1) |
| N822K | 2466T > G | 0.018 | ||||
| 10 | R/R | D816V | 2447A > T | 0.234 | None | t(8;21)(q22;q22.1) |
| 39 | R/R | D816V | 2447A > T | 0.252 | RAD21 | t(8;21)(q22;q22.1) |
| D816Y | 2446G > T | 0.056 | ||||
| 45 | R/R | D816Y | 2446G > T | 0.923 | CD36 | t(8;21)(q22;q22.1) |
| 76 | R/R | D816V | 2447A > T | 0.932 | NF1 | t(8;21)(q22;q22.1) |
| 94 | R/R | D816V | 2447A > T | 0.459 | RAD21, NPM1 | Normal |
| 111 | R/R | D816V | 2447A > T | 0.338 | SETD2 | 3q Abnormality |
| 121 | R/R | D816Y | 2446G > T | 0.021 | CBL | inv(16)/t(16;16) |
| 146 | R/R | D816V | 2447A > T | 0.082 | GATA2, HIST1H2BJ | t(8;21)(q22;q22.1) |
| 175 | R/R | N822K | 2466T > G | 0.461 | GATA2, PHF6, ATM | t(8;21)(q22;q22.1) |
| Drug | Primary Targets | FDA-Approved Disease |
|---|---|---|
| Imatinib | BCR-ABL1 | CML, Ph+ALL, HES, GIST, SM, DFSP |
| Dasatinib | BCR-ABL1 | CML, PhALL |
| Sunitinib | VEGFR and FLT3 | GIST, RCC, Pancreatic Cancer |
| Regorafenib | VEGFR | GIST, HCC, Colorectal Cancer |
| Midostaurin | FLT3 | AML (FLT3 mutation), SM |
| Ripretinib | KIT | GIST |
| Avapritinib | KIT/PDGFRA | GIST, SM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katagiri, S.; Chi, S.; Minami, Y.; Fukushima, K.; Shibayama, H.; Hosono, N.; Yamauchi, T.; Morishita, T.; Kondo, T.; Yanada, M.; et al. Mutated KIT Tyrosine Kinase as a Novel Molecular Target in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2022, 23, 4694. https://doi.org/10.3390/ijms23094694
Katagiri S, Chi S, Minami Y, Fukushima K, Shibayama H, Hosono N, Yamauchi T, Morishita T, Kondo T, Yanada M, et al. Mutated KIT Tyrosine Kinase as a Novel Molecular Target in Acute Myeloid Leukemia. International Journal of Molecular Sciences. 2022; 23(9):4694. https://doi.org/10.3390/ijms23094694
Chicago/Turabian StyleKatagiri, Seiichiro, SungGi Chi, Yosuke Minami, Kentaro Fukushima, Hirohiko Shibayama, Naoko Hosono, Takahiro Yamauchi, Takanobu Morishita, Takeshi Kondo, Masamitsu Yanada, and et al. 2022. "Mutated KIT Tyrosine Kinase as a Novel Molecular Target in Acute Myeloid Leukemia" International Journal of Molecular Sciences 23, no. 9: 4694. https://doi.org/10.3390/ijms23094694