
Characterization of the Complete Mitochondrial Genome of the Elongate Loach and Its Phylogenetic Implications in Cobitidae
 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Mitochondrial Genome Annotation and Analysis
2.3. Phylogenetic Analyses
3. Results and Discussion
3.1. Mitochondrial Structural Characteristics
3.2. Protein Coding Genes
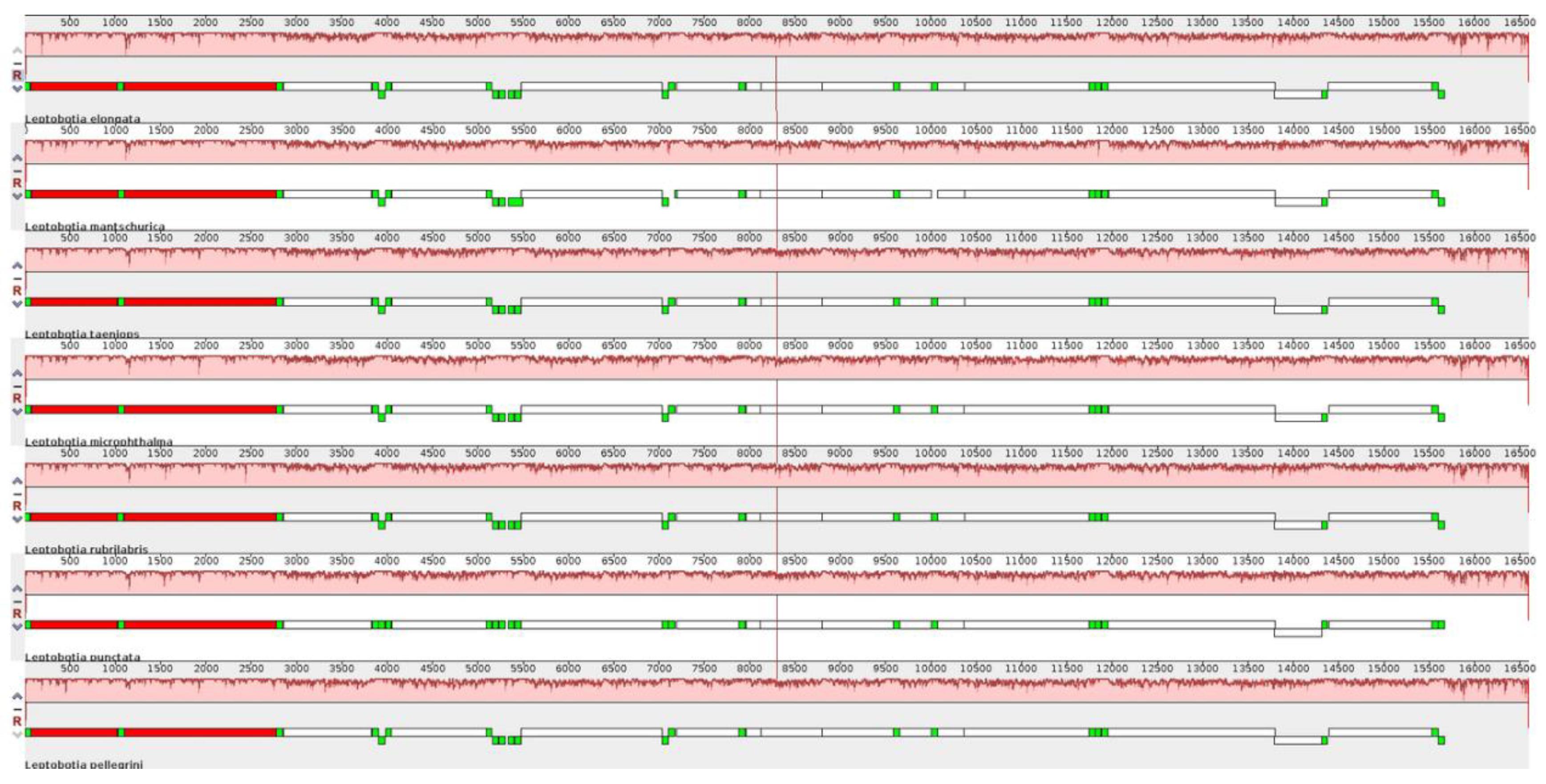
3.3. Genome Comparative Analysis
3.4. Ribosomal RNA and Transfer RNA Genes
3.5. Non-Coding Regions
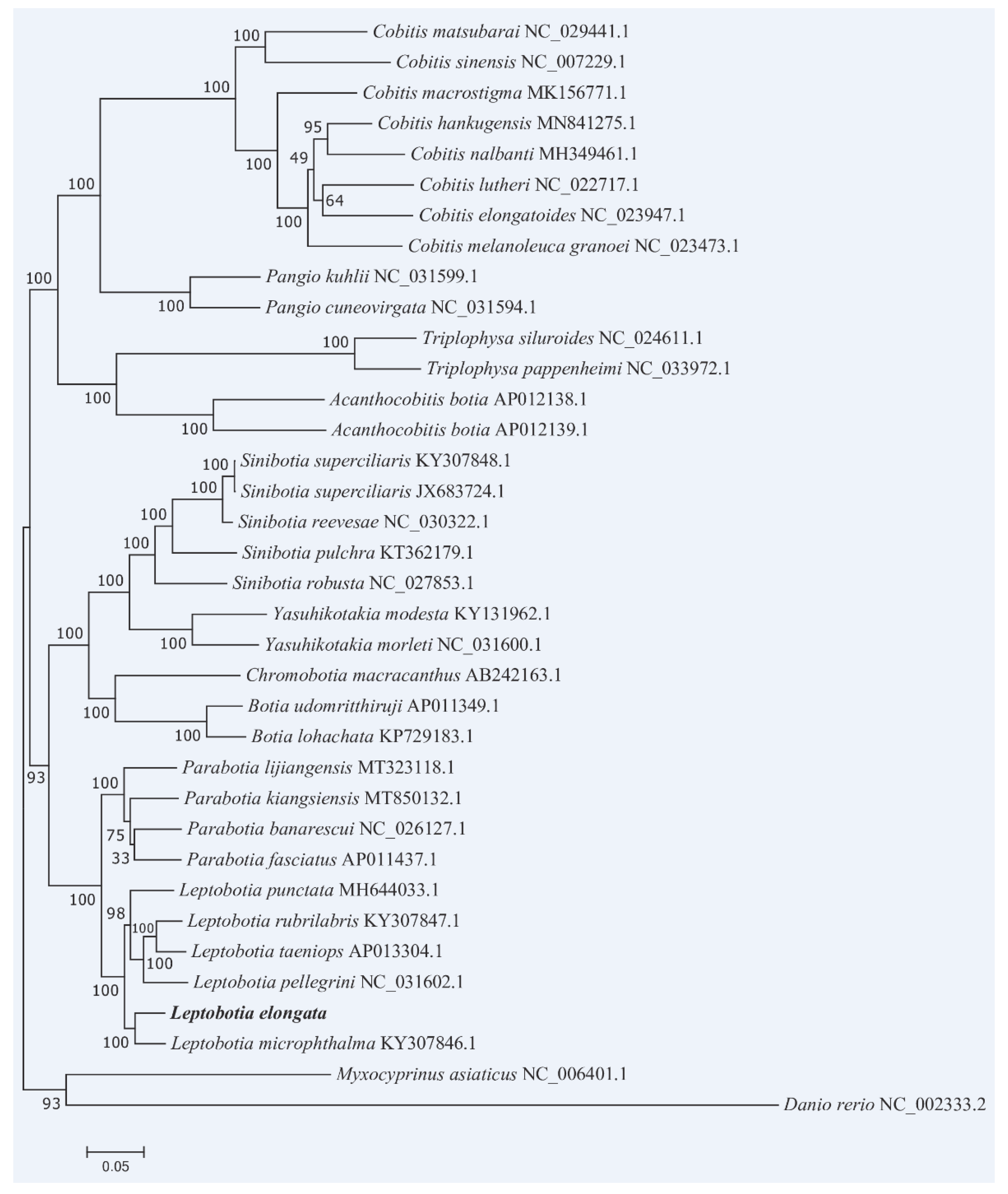
3.6. Phylogenetic Relationships
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yuan, Y.C.; Yang, H.J.; Gong, S.Y.; Liang, Y.Q. Threatened fishes of the world: Leptobotia elongata Bleeker, 1870 (Bottiinae). Environ. Biol. Fishes 2010, 87, 295–296. [Google Scholar] [CrossRef]
- Chen, K.G.; Wang, Z.J.; Yue, X.J. Study of the structurs of the digestive system in Leptobotia elongata. Southwest Agric. Univ. 2002, 24, 1000–2642. [Google Scholar] [CrossRef]
- Tian, H.W.; Duan, X.B.; Xiong, X.; Luo, H.Q.; Liu, S.P.; Chen, D.Q. Estimation of growth and population parameters of elongate loach (Leptobotia elongata) in the upper reaches of the Yangtze River. Yangtze Basin 2013, 22, 1305–1312. [Google Scholar]
- Zhang, Y.; Cao, X.; Zou, Y.; Yan, Z.; Huang, Y.; Zhu, Y.; Gao, J. De novo gonad transcriptome analysis of elongate loach (Leptobotia elongata) provides novel insights into sex-related genes. Comp. Biochem. Physiol. Part D 2022, 42, 100962. [Google Scholar] [CrossRef]
- Yue, P.; Chen, Y. China Red Data Book of Endangered Animals: Pisces; Science Press: Beijing, China, 1998; pp. 240–243. [Google Scholar]
- Regan, C.T. The classification of the teleostean fishes of the order Ostariophysi. I. Cyprinidae. Ann. Mag. Nat. Hist. 1911, 8, 13–32. [Google Scholar] [CrossRef]
- Hora, S.L. Classification, bionomics and evolution of homalopterid fishes. Mem. Indian Mus. 1932, 12, 263–330. [Google Scholar]
- Berg, L.S. Classification of fishes both recent and fossil. Science 1940, 107, 87–345. [Google Scholar] [CrossRef]
- Liu, H.Z.; Tzeng, C.S.; Teng, H.Y. Sequence variations in the mitochondrial DNA control region and their implications for the phylogeny of the Cypriniformes. Can. J. Zool. 2002, 80, 569–581. [Google Scholar] [CrossRef]
- Nalbant, T.T. Sixty million years of evolution. Part one: Family Botiidae (Pisces: Ostariophysi: Cobitoidea). Trav. Mus. Hist. Nat. “Grigore Antipa” 2002, 44, 309–344. [Google Scholar]
- Liu, H.Y.; Cai, J.; Xie, Z.G.; Xiong, F.; Wang, Y.; Wang, Q.; Yu, J.X.; Zhai, D.D.; Xia, M.; Chen, Y.Y. DNA Barcodes for species identification and systematic evolution of cobitidae fish. Acta Agric. Univ. Jiangxiensis 2020, 42, 766–777. [Google Scholar] [CrossRef]
- Liu, S.Q.; Mayden, R.L.; Zhang, J.B.; Yu, D.; Tang, Q.Y.; Deng, X.; Liu, H.Z. Phylogenetic relationships of the Cobitoidea (Teleostei: Cypriniformes) inferred from mitochondrial and nuclear genes with analyses of gene evolution. Gene 2012, 508, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.Y.; Liu, H.Z.; Mayden, R.; Xiong, B.X. Comparison of evolutionary rates in the mitochondrial DNA cytochrome b gene and control region and their implications for phylogeny of the Cobitoidea (Teleostei: Cypriniformes). Mol. Phylogenetics Evol. 2006, 39, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Wang, Y.; Liang, R.; Feng, J.; Kefeng, A. Field observations of the lethality characteristics of endangered and endemic fish under the stress of total dissolved gas supersaturation. River Res. Appl. 2020, 38, 1156–1167. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Ding, Y.; Zheng-Xuan, G.U.; Huang, X.X.; Wen, Y.Z.; Min, S. Artificial propagation and embryonic development observation of Leptobotia elongata from Jinsha River and Yangtze River. Hubei Agric. Sci. 2018, 57, 104–107. [Google Scholar] [CrossRef]
- Liu, D.; Li, X.; Song, Z. No decline of genetic diversity in elongate loach (Leptobotia elongata) with a tendency to form population structure in the upper Yangtze River. Glob. Ecol. Conserv. 2020, 23, e01072. [Google Scholar] [CrossRef]
- Liu, D.Q.; Wu, J.Y.; Deng, L.J.; Gan, W.X.; Du, L.M.; Song, Z.B. Developmentof Microsatellite Markers for Leptobotia elongata (Cypriniformes: Cobitidae) Using 454 Sequencing and Cross-species Amplification. Pak. J. Zool. 2014, 4, 1147–1151. [Google Scholar]
- Li, P.; Yang, C.Z.; Tu, F.Y.; Liu, G.X. The complete mitochondrial genome of the Elongate loach Leptobotia elongata (Cypriniformes: Cobitidae). Mitochondrial DNA 2012, 23, 352–354. [Google Scholar] [CrossRef]
- Caccone, A.; Gentile, G.; Burns, C.E.; Sezzi, E.; Powell, J.R. Extreme difference in rate of mitochondrial and nuclear DNA evolution in a large ectotherm, Galápagos tortoises. Mol. Phylogenetics Evol. 2004, 31, 794–798. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, P.D.; Zhang, D.Z.; Zhang, H.B.; Tang, B.P.; Liu, Q.N. Mitochondrial genome of the yellow catfish Pelteobagrus fulvidraco and insights into Bagridae phylogenetics. Genomics 2018, 111, 1258–1265. [Google Scholar] [CrossRef]
- Wang, C.H.; Chen, Q.; Lu, G.Q.; Xu, J.W.; Yang, Q.L.; Li, S. Complete mitochondrial genome of the grass carp (Ctenopharyngodon idella, Teleostei): Insight into its phylogenic position within Cyprinidae. Gene 2008, 424, 96–101. [Google Scholar] [CrossRef]
- Tang, K.L.; Agnew, M.K.; Hirt, M.V.; Sado, T.Y.; Schneider, L.M.; Freyhof, J.; Sulaiman, Z.; Swartz, E.; Vidthayanon, C.; Miya, M.; et al. Systematics of the subfamily Danioninae (Teleostei: Cypriniformes: Cyprinidae). Mol. Phylogenetics Evol. 2010, 57, 189–214. [Google Scholar] [CrossRef] [PubMed]
- Muniyangd, N.; Raja, M.; Vikram, P. Genetic characterization of Bagarius species using cytochrome c oxidase I and cytochrome b genes. Mitochondrial DNA 2016, 27, 3781–3783. [Google Scholar] [CrossRef]
- Chen, D.X.; Chu, W.Y.; Liu, X.L.; Nong, X.X.; Li, Y.L.; Du, S.J.; Zhang, J. Phylogenetic studies of three sinipercid fishes (Perciformes: Sinipercidae) based on complete mitochondrial DNA sequences. Mitochondrial DNA 2012, 23, 70–76. [Google Scholar] [CrossRef]
- Pavan-Kumar, A.; Raman, S.; Koringa, P.G.; Patel, N.; Chaudhari, A. Complete mitochondrial genome of threatened mahseer Tor tor (Hamilton 1822) and its phylogenetic relationship within Cyprinidae family. J. Genet. 2016, 95, 853–863. [Google Scholar] [CrossRef]
- Zou, X.H.; Ge, S. Conflicting gene trees and phylogenomics. J. Syst. Evol. 2008, 46, 795–807. [Google Scholar] [CrossRef]
- Rieppel, O. ‘Total evidence’ in phylogenetic systematics. Biol. Philos. 2009, 24, 607–622. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 77–455. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 74–267. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 80–772. [Google Scholar] [CrossRef]
- Wang, D.P.; Zhang, Y.B.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinf. 2010, 8, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Conrad, I.; Craft, A.; Thurman, C.L.; Baeza, J.A. The complete mitochondrial genome of the red-jointed brackish-water fiddler crab Minuca minax (LeConte 1855) (Brachyura: Ocypodidae): New family gene order, and purifying selection and phylogenetic informativeness of protein coding genes. Genomics 2021, 113, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P.; Grant, R.J.; Domselaar, G.V. Visualizing and comparing circular genomes using the CGView family of tools. Brief. Bioinform. 2019, 20, 1576–1582. [Google Scholar] [CrossRef]
- Zhong, L.Q.; Wang, M.H.; Li, D.; Tang, S.; Zhang, T.Q.; Bian, W.J.; Chen, X.H. Complete mitochondrial genome of Odontobutis haifengensis (Perciformes, Odontobutiae): A unique rearrangement of tRNAs and additional non-coding regions identified in the genus Odontobutis. Genomics 2018, 110, 382–388. [Google Scholar] [CrossRef]
- Prabhu, V.R.; Singha, H.S.; Kumar, R.G.; Gopalakrishnan, A.; Nagarajan, M. Characterization of the complete mitochondrial genome of Barilius malabaricus and its phylogenetic implications. Genomics 2020, 112, 2154–2163. [Google Scholar] [CrossRef]
- Wei, S.; Shi, M.; Chen, X.; Sharkey, M.; Achterberg, C.; Ye, G.; He, J. New views on strand asymmetry in insect mitochondrial genomes. PLoS ONE 2010, 5, e12708. [Google Scholar] [CrossRef]
- Tian, H.; Wang, D.; Jia, X.; Duan, X.; Chen, D. The mitogenome of Leptobotia microphthalma (Teleostei, Cypriniformes: Cobitidae). Mitochondrial DNA 2014, 25, 261–262. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Li, Y.; Liu, Z.; Chen, Q.; Shen, Y. The complete mitochondrial genome of Cobitis macrostigma (Cypriniformes: Cobitidae: Cobitinae) and a phylogenetic implication for its closely related species. Biologia 2020, 75, 393–399. [Google Scholar] [CrossRef]
- Ma, Q.; Zhang, T.L.; Chen, L.; Tang, Q.Y. The Complete Mitochondrial Genomes of Parabotia Kiangsiensis (Cypriniformes: Botiidae). Mitochondrial DNA 2020, 5, 3629–3631. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Garey, J.R.; Wolstenholme, D.R. Platyhelminth mitochondrial DNA: Evidence for early evolutionary origin of a tRNAserAGN that contains a dihydrouridine arm replacement loop, and of serine-specifying AGA and AGG codons. J. Mol. Evol. 1989, 28, 374–387. [Google Scholar] [CrossRef]
- Sharma, A.; Siva, C.; Ali, S.; Sahoo, P.K.; Sarma, D. The complete mitochondrial genome of the medicinal fish, Cyprinion semiplotum: Insight into its structural features and phylogenetic implications. Int. J. Biol. Macromol. 2020, 164, 939–948. [Google Scholar] [CrossRef]
- Lee, Y.S.; Prakash Patil, M.; Kim, J.O.; Lee, Y.J.; Seo, Y.B.; Kim, J.K.; Suryawanshi, R.K. The Complete Mitochondrial Genome of the Fivespot Flounder, Pseudorhombus pentophthalmus (Pleuronectiformes: Paralichthyidae), from Korea and Its Phylogenetic Analysis. Fishes 2023, 8, 150. [Google Scholar] [CrossRef]
- Pavan-Kumar, A.; Singh, S.; Mishra, A.; Suman, S.; Gireesh-Babu, P.; Chaudhari, A.; Shen, K.N.; Borsa, P. Characterization of mitochondrial genome of Indian Ocean blue-spotted maskray, Neotrygon indica and its phylogenetic relationship within Dasyatidae Family. Int. J. Biol. Macromol. 2022, 223, 458–467. [Google Scholar] [CrossRef] [PubMed]
- William, B. Codon distribution in vertebrate genes may be used to predict gene length. J. Mol. Biol. 1987, 197, 379–388. [Google Scholar] [CrossRef]
- Sueoka, N. Two Aspects of DNA Base Composition: G+C Content and Translation-Coupled Deviation from Intra-Strand Rule of A=T and G=C. J. Mol. Evol. 1999, 49, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Akashi, H. Translational selection and yeast proteome evolution. Genetics 2003, 164, 1291–1303. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, G.; Bernardi, G. Compositional Constraints and Genome Evolution. J. Mol. Evol. 1986, 24, 1–11. [Google Scholar] [CrossRef] [PubMed]
- D’Onofrio, G.; Mouchiroud, D.; Aïssani, B.; Gautier, C.; Bernardi, G. Correlations between the compositional properties of human genes, codon usage, and amino acid composition of proteins. J. Mol. Evol. 1991, 32, 504–510. [Google Scholar] [CrossRef]
- Foster, P.G.; Jermiin, L.S.; Hickey, D.A. Nucleotide Composition Bias Affects Amino Acid Content in Proteins Coded by Animal Mitochondria. J. Mol. Evol. 1997, 44, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Dudgeon, C.L.; Broderick, D.; Ovenden, J.R. IUCN classification zones concord with, but underestimate, the population genetic structure of the zebra shark Stegostoma fasciatum in the Indo-West Pacific. Mol. Ecol. 2009, 18, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486–487. [Google Scholar] [CrossRef]
- Yang, Z.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chen, Y.; Yan, J.; Page, L.M. Phylogenetic analysis and osteological comparison of the cave-dwelling spined loach, Bibarba parvoculus (Cypriniformes: Cobitidae), and its surface congener. Zool. J. Linn. Soc. 2021, 191, 1059–1074. [Google Scholar] [CrossRef]
- Shen, Y.J.; Wang, J.; Zhang, F.B. Complete Mitochondrial Genome of Parabotia bimaculata (Cypriniformes: Cobitidae: Botiinae), an Endemic Riverine Loach in China and Phylogenetic Analysis for Botiinae. Thalassas 2020, 36, 387–393. [Google Scholar] [CrossRef]
- Yang, X.G.; Lian, Y.X.; Chen, M.M.; Li, X.Q.; Yu, D.P. Characterization and phylogenetic analysis of the complete mitochondrial genome of sun loach (Yasuhikotakia eos). Mitochondrial DNA 2021, 6, 13–14. [Google Scholar] [CrossRef]
- Baeza, J.A. The complete mitochondrial genome of the Caribbean spiny lobster Panulirus argus. Sci. Rep. 2018, 8, 17690. [Google Scholar] [CrossRef]
- Inoue, J.G.; Miya, M.; Tsukamoto, K.; Nishida, M. Complete mitochondrial DNA sequence of the Japanese sardine Sardinops melanostictus. Fish. Sci. 2010, 66, 924–932. [Google Scholar] [CrossRef]
- Varani, G.; McClain, W.H. The G·U wobble base pair: A fundamental building block of RNA structure crucial to RNA function in diverse biological systems. EMBO Rep. 2000, 1, 18–23. [Google Scholar] [CrossRef]
- Chen, L.; Lin, Y.F.; Xiao, Q.; Lin, Y.; Du, Y.; Lin, C.X.; Ward-Fear, G.; Hu, C.C.; Qu, Y.F.; Li, H. Characterization of the complete mitochondrial genome of the many-lined sun skink (Eutropis multifasciata) and comparison with other Scincomorpha species. Genomics 2021, 113, 2526–2536. [Google Scholar] [CrossRef]
- Gao, Y.C.; Zhang, J.; Wang, Q.H.; Liu, Q.N.; Tang, B.P. The Complete Mitochondrial Genome of Box Tree Moth Cydalima perspectalis and Insights into Phylogenetics in Pyraloidea. Animals 2023, 13, 1045. [Google Scholar] [CrossRef]
- Wang, X.; Wang, J.; He, S.; Mayden, R.L. The complete mitochondrial genome of the Chinese hook snout carp Opsariichthys bidens (Actinopterygii: Cypriniformes) and an alternative pattern of mitogenomic evolution in vertebrate. Gene 2007, 399, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Broughton, R.E.; Milam, J.E.; Roe, B.A. The Complete Sequence of the Zebrafish (Danio rerio) Mitochondrial Genome and Evolutionary Patterns in Vertebrate Mitochondrial DNA. Genome Res. 2001, 11, 1958–1967. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.W. Study on Botoid Fishes of China. Sinensia 1936, 7, 1–49. [Google Scholar]
- Tang, Q.Y. Molecular phylogeny of the cobitoidea. HZAU 2005. [Google Scholar] [CrossRef]
- Slechtova, V.; Bohlen, J.; Freyhof, J.; Rab, P. Molecular phylogeny of the Southeast Asian freshwater fish family Botiidae (Teleostei: Cobitoldea) and the origin of polyploidy in their evolution. Mol. Phylogenetics Evol. 2006, 39, 529–541. [Google Scholar] [CrossRef]
- Tang, Q.Y.; Yu, D.; Liu, H.Z. Leptobotia zebra Should Be Revised as Sinibotia zebra (Cypriniformes: Botiidae). Zool. Res. 2008, 29, 1–9. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Genus | Species | Assession Number |
|---|---|---|---|
| Myxocyprinae | Myxocyprinus | Myxocyprinus asiaticus | NC_006401.1 |
| Cyprinidae | Danio | Danio rerio | NC_002333.2 |
| Cobitidae | Leptobotia | Leptobotia microphthalma | KY307846.1 |
| Leptobotia | Leptobotia elongata | OR818399 | |
| Leptobotia | Leptobotia pellegrini | NC_031602.1 | |
| Leptobotia | Leptobotia taeniops | AP013304.1 | |
| Leptobotia | Leptobotia rubrilabris | KY307847.1 | |
| Leptobotia | Leptobotia punctata | MH644033.1 | |
| Leptobotia | Leptobotia mantschurica | AB242170.1 | |
| Parabotia | Parabotia fasciata | AP011437.1 | |
| Parabotia | Parabotia banarescui | NC_026127.1 | |
| Parabotia | Parabotia kiangsiensis | MT850132.1 | |
| Parabotia | Parabotia lijiangensis | MT323118.1 | |
| Botia | Botia udomritthiruji | AP011349.1 | |
| Botia | Botia lohachata | KP729183.1 | |
| Chromobotia | Chromobotia macracanthus | AB242163.1 | |
| Yasuhikotakia | Yasuhikotakia morleti | NC_031600.1 | |
| Yasuhikotakia modesta | KY131962.1 | ||
| Sinibotia | Sinibotia robusta | NC_027853.1 | |
| Sinibotia pulchra | KT362179.1 | ||
| Sinibotia reevesae | NC_030322.1 | ||
| Sinibotia superciliaris | JX683724.1 | ||
| Sinibotia superciliaris | KY307848.1 | ||
| Acanthocobitis | Acanthocobitis botia | AP012139.1 | |
| Acanthocobitis botia | AP012138.1 | ||
| Triplophysa | Triplophysa pappenheimi | NC_033972.1 | |
| Triplophysa siluroides | NC_024611.1 | ||
| Pangio | Pangio kuhlii | NC_031599.1 | |
| Pangio cuneovirgata | NC_031594.1 | ||
| Cobitis | Cobitis lutheri | NC_022717.1 | |
| Cobitis melanoleuca granoei | NC_023473.1 | ||
| Cobitis nalbanti | MH349461.1 | ||
| Cobitis elongatoides | NC_023947.1 | ||
| Cobitis hankugensis | MN841275.1 | ||
| Cobitis macrostigma | MK156771.1 | ||
| Cobitis sinensis | NC_007229.1 | ||
| Cobitis matsubarai | NC_029441.1 |
| L. elongata | Size (bp) | A% | T% | G% | C% | A + T% | G + C% | AT-Skew | GC-Skew |
|---|---|---|---|---|---|---|---|---|---|
| Mitogenome | 16,591 | 30.79 | 24.77 | 16.17 | 28.27 | 55.56 | 44.44 | 0.108 | −0.272 |
| PCGs | 11,430 | 28.56 | 26.77 | 15.55 | 29.13 | 55.33 | 44.67 | 0.032 | −0.304 |
| tRNAs | 1558 | 28.18 | 25.8 | 23.49 | 22.53 | 53.98 | 46.02 | 0.044 | 0.021 |
| rRNAs | 2638 | 34.04 | 19.48 | 21.04 | 25.44 | 53.53 | 46.47 | 0.272 | −0.095 |
| Dloop | 926 | 35.64 | 31.75 | 13.71 | 18.9 | 67.39 | 32.61 | 0.058 | −0.159 |
| Position | Codon | ||||||
|---|---|---|---|---|---|---|---|
| Gene | Stand | From | To | Size | Intergenic Length | Start | Stop |
| tRNA-phe | H | 1 | 69 | 69 | 0 | ||
| 12 S rRNA | H | 70 | 1024 | 955 | 0 | ||
| tRNA-val | H | 1025 | 1096 | 72 | 0 | ||
| 16 S rRNA | H | 1097 | 2779 | 1683 | 0 | ||
| tRNA-leu | H | 2780 | 2854 | 75 | 0 | ||
| nd1 | H | 2855 | 3829 | 975 | 0 | ATG | TAA |
| tRNA-ile | H | 3838 | 3909 | 72 | 8 | ||
| tRNA-gln | L | 3908 | 3978 | 71 | −2 | ||
| tRNA-met | H | 3980 | 4048 | 69 | 1 | ||
| nd2 | H | 4049 | 5094 | 1046 | 0 | ATG | TA- |
| tRNA-trp | H | 5095 | 5163 | 69 | 0 | ||
| tRNA-ala | L | 5166 | 5234 | 69 | 2 | ||
| tRNA-asn | L | 5236 | 5308 | 73 | 1 | ||
| OL | L | 5310 | 5340 | 39 | 1 | ||
| tRNA-cys | L | 5339 | 5404 | 66 | −2 | ||
| tRNA-tyr | L | 5406 | 5476 | 71 | 1 | ||
| cox1 | H | 5478 | 7028 | 1551 | 1 | GTG | TAA |
| tRNA-ser | L | 7030 | 7100 | 71 | 1 | ||
| tRNA-asp | H | 7103 | 7174 | 72 | 2 | ||
| cox2 | H | 7188 | 7878 | 691 | 13 | ATG | T-- |
| tRNA-lys | H | 7879 | 7954 | 76 | 0 | ||
| ATPase8 | H | 7956 | 8123 | 168 | 1 | ATG | TAA |
| ATPase6 | H | 8114 | 8797 | 684 | −10 | ATG | TAA |
| cox3 | H | 8797 | 9581 | 785 | 1 | ATG | TA- |
| tRNA-gly | H | 9582 | 9653 | 72 | 0 | ||
| nd3 | H | 9654 | 10,002 | 349 | 0 | ATG | T-- |
| tRNA-arg | H | 10,003 | 10,072 | 70 | 0 | ||
| nd4l | H | 10,073 | 10,369 | 297 | 0 | ATG | TAA |
| nd4 | H | 10,363 | 11,744 | 1382 | −7 | ATG | TA- |
| tRNA-his | H | 11,745 | 11,814 | 70 | 0 | ||
| tRNA-ser | H | 11,815 | 11,881 | 67 | 0 | ||
| tRNA-leu | H | 11,883 | 11,955 | 73 | 1 | ||
| nd5 | H | 11,956 | 13,794 | 1839 | 0 | ATG | TAA |
| nd6 | L | 13,791 | 14,312 | 522 | −4 | ATG | TAA |
| tRNA-glu | L | 14,313 | 14,381 | 69 | 0 | ||
| cytb | H | 14,386 | 15,526 | 1141 | 4 | ATG | T-- |
| tRNA-thr | H | 15,527 | 15,598 | 72 | 0 | ||
| tRNA-pro | L | 15,597 | 15,666 | 70 | −2 | ||
| CR | H | 15,666 | 16,591 | 926 | 0 | ||
| Codon | No. | RSCU | Codon | No. | RSCU | Codon | No. | RSCU |
|---|---|---|---|---|---|---|---|---|
| UAA() | 7 | 1 | AAA(K) | 38 | 1.8536 | CGG(R) | 5 | 0.5 |
| GCA(A) | 68 | 1.4468 | AAG(K) | 3 | 0.1464 | CGU(R) | 5 | 0.5 |
| GCC(A) | 94 | 2 | CUA(L) | 123 | 2.271 | AGC(S) | 24 | 1.161 |
| GCG(A) | 3 | 0.064 | CUC(L) | 57 | 1.0524 | AGU(S) | 4 | 0.1938 |
| GCU(A) | 23 | 0.4892 | CUG(L) | 22 | 0.4062 | UCA(S) | 50 | 2.4192 |
| UGC(C) | 7 | 1.1666 | CUU(L) | 53 | 0.9786 | UCC(S) | 22 | 1.0644 |
| UGU(C) | 5 | 0.8334 | UUA(L) | 51 | 0.9414 | UCG(S) | 1 | 0.0486 |
| GAC(D) | 25 | 1.3158 | UUG(L) | 19 | 0.351 | UCU(S) | 23 | 1.113 |
| GAU(D) | 13 | 0.6842 | AUA(M) | 60 | 2.0226 | ACA(T) | 72 | 1.87 |
| GAA(E) | 36 | 1.44 | AUG(M) | 28 | 0.9438 | ACC(T) | 59 | 1.5324 |
| GAG(E) | 14 | 0.56 | GUG(M) | 1 | 0.0336 | ACG(T) | 1 | 0.026 |
| UUC(F) | 63 | 0.9618 | AAC(N) | 54 | 1.4594 | ACU(T) | 22 | 0.5716 |
| UUU(F) | 68 | 1.0382 | AAU(N) | 20 | 0.5406 | GUA(V) | 52 | 1.5524 |
| GGA(G) | 48 | 1.4116 | CCA(P) | 47 | 1.6348 | GUC(V) | 23 | 0.6864 |
| GGC(G) | 31 | 0.9116 | CCC(P) | 47 | 1.6348 | GUG(V) | 24 | 0.7164 |
| GGG(G) | 39 | 1.1472 | CCG(P) | 6 | 0.2088 | GUU(V) | 35 | 1.0448 |
| GGU(G) | 18 | 0.5296 | CCU(P) | 15 | 0.5216 | UGA(W) | 45 | 1.6364 |
| CAC(H) | 42 | 1.7142 | CAA(Q) | 45 | 1.9148 | UGG(W) | 10 | 0.3636 |
| CAU(H) | 7 | 0.2858 | CAG(Q) | 2 | 0.0852 | UAC(Y) | 26 | 0.963 |
| AUC(I) | 59 | 0.792 | CGA(R) | 23 | 2.3 | UAU(Y) | 28 | 1.037 |
| AUU(I) | 90 | 1.208 | CGC(R) | 7 | 0.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ke, Z.; Zhou, K.; Hou, M.; Luo, H.; Li, Z.; Pan, X.; Zhou, J.; Jing, T.; Ye, H. Characterization of the Complete Mitochondrial Genome of the Elongate Loach and Its Phylogenetic Implications in Cobitidae. Animals 2023, 13, 3841. https://doi.org/10.3390/ani13243841
Ke Z, Zhou K, Hou M, Luo H, Li Z, Pan X, Zhou J, Jing T, Ye H. Characterization of the Complete Mitochondrial Genome of the Elongate Loach and Its Phylogenetic Implications in Cobitidae. Animals. 2023; 13(24):3841. https://doi.org/10.3390/ani13243841
Chicago/Turabian StyleKe, Zhenlin, Kangqi Zhou, Mengdan Hou, Hui Luo, Zhe Li, Xianhui Pan, Jian Zhou, Tingsen Jing, and Hua Ye. 2023. "Characterization of the Complete Mitochondrial Genome of the Elongate Loach and Its Phylogenetic Implications in Cobitidae" Animals 13, no. 24: 3841. https://doi.org/10.3390/ani13243841