
Comprehensive Analysis of Phylogenetic Relationship and Optimal Codons in Mitochondrial Genomes of the Genus Pseudogastromyzon

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples Collection
2.2. DNA Extraction, PCR Amplification, and Sequencing
2.3. Complete Mitogenome Analysis
2.4. Determination of Optimal Codons and Cluster Analysis
2.5. Phylogenetic Analysis
2.6. Divergence Times Estimation
3. Results and Discussion
3.1. Mitochondrial Genome Organization and Base Composition
3.2. Protein-Coding Genes
3.3. Transfer and Ribosomal RNA Genes
3.4. Noncoding Regions
3.5. Mitochondrial Gene Rearrangement
3.6. Screening of Optimal Codons and Cluster Analysis
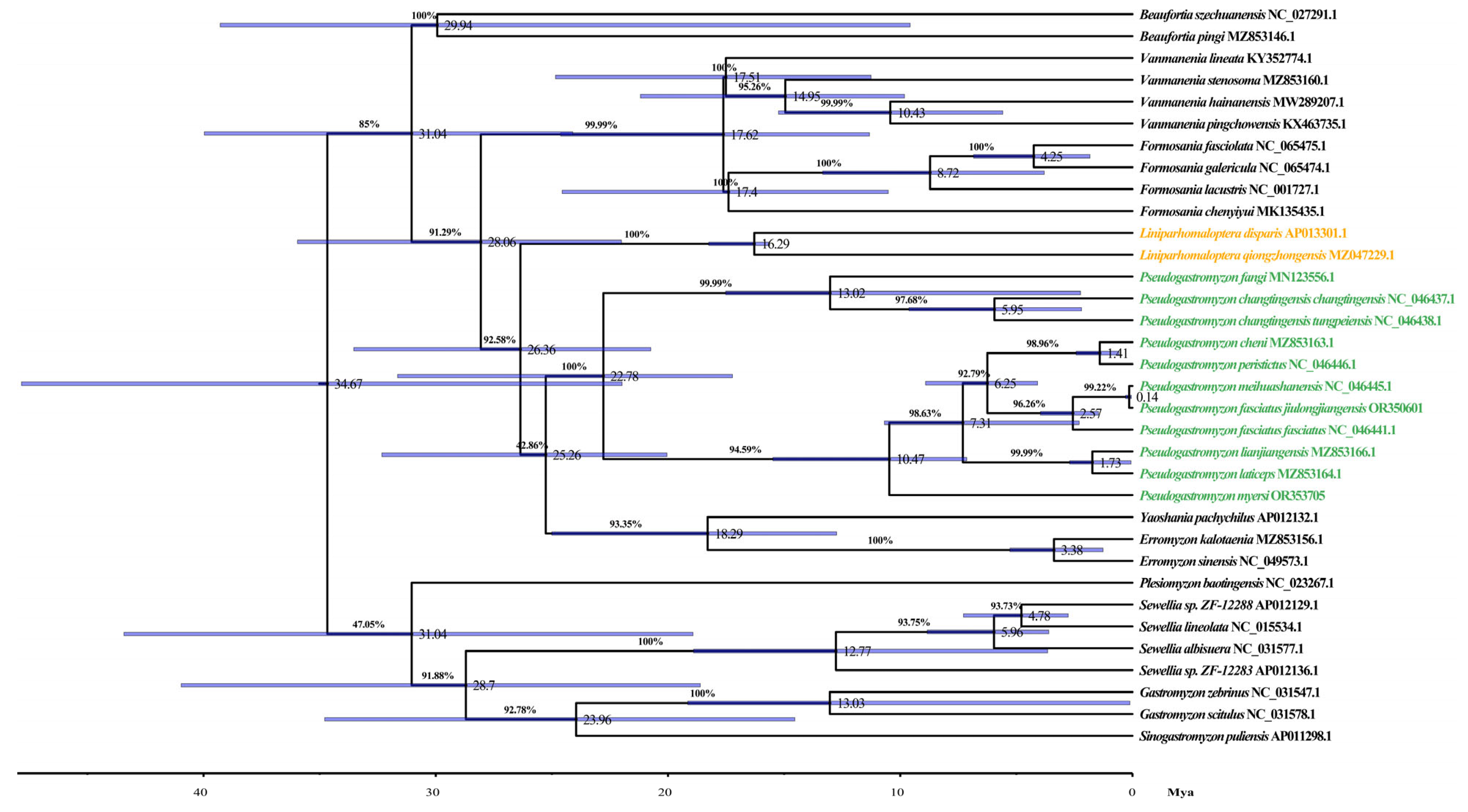
3.7. Phylogenetic Analysis and Divergence Time Estimation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- He, M.; Yuan, D.; Cui, L.; Zou, L.; Jiang, Y. Study on fish community and impact factors in Mastacembelus aculedtus National Aquatic Reserve in Tingjiang River. J. Shanghai Ocean. Univ. 2015, 24, 121–129. [Google Scholar]
- Chen, J.; Chen, Y.; Tang, W.; Lei, H.; Yang, J.; Song, X. Resolving phylogenetic relationships and taxonomic revision in the Pseudogastromyzon (Cypriniformes, Gastromyzonidae) genus: Molecular and morphological evidence for a new genus, Labigastromyzon. Integr. Zool. 2023. early view. [Google Scholar] [CrossRef]
- Yang, G.Y.; Dudgeon, D. Dietary variation and food selection by an algivorous loach (Pseudogastromyzon myersi: Balitoridae) in Hong Kong streams. Mar. Freshw. Res. 2010, 61, 49–56. [Google Scholar] [CrossRef]
- Song, X.; Yang, Y.; Tang, W. Complete mitochondrial genome of Pseudogastromyzon fasciatus (Osteichthyes: Gastromyzontidae). Mitochondrial DNA Part B Resour. 2016, 1, 686–687. [Google Scholar] [CrossRef]
- Zheng, C.; Li, J. Fish species of the genus Pseudogastromyzon in China. J. Jinan Univ. (Nat. Sci. Med. Ed.) 1986, 1, 75–84. [Google Scholar]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J.; Zody, M.C.; et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Wolstenholme, D.R. Genetic novelties in mitochondrial genomes of multicellular animals. Curr. Opin. Genet. Dev. 1992, 2, 918–925. [Google Scholar] [CrossRef]
- Clayton, D.A. Replication and transcription of vertebrate mitochondrial DNA. Annu. Rev. Cell Biol. 1991, 7, 453–478. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, S.; Tian, Z.; Wang, D.; Xu, S. Identification of optimal codons and analysis of phylogenetic relationship in Osteochilus salsburyi (Teleostei: Cypriniformes) based on complete mitogenome. Front. Ecol. Evol. 2023, 11, 1170927. [Google Scholar] [CrossRef]
- Ingman, M.; Gyllensten, U. mtDB: Human Mitochondrial Genome Database, a resource for population genetics and medical sciences. Nucleic Acids Res. 2006, 34, D749–D751. [Google Scholar] [CrossRef]
- Lau, N.-S.; Sam, K.-K.; Ahmad, A.B.; Siti, K.-A.; Ahmad Zafir, A.W.; Shu-Chien, A.C. Gene Arrangement and Adaptive Evolution in the Mitochondrial Genomes of Terrestrial Sesarmid Crabs Geosesarma faustum and Geosesarma penangensis. Front. Ecol. Evol. 2021, 9, 778570. [Google Scholar] [CrossRef]
- Yang, Y.; Song, X.; Tang, W. Complete mitochondrial DNA sequence of Pseudogastromyzon changtingensis (Osteichthyes: Gastromyzontidae). Mitochondrial DNA Part B Resour. 2016, 1, 871–872. [Google Scholar] [CrossRef]
- Lv, Y.; Zhang, K.; Liu, Z.; Hu, Z.; Wang, K.; Zhou, H. Complete mitochondrial genome of the Pseudogastromyzon jiulongjiangensis Chen (Cypriniformes, Homalopteridae) and phylogenetic analysis of the Cyprinoidei. Mitochondrial DNA Part A 2016, 27, 2658–2660. [Google Scholar] [CrossRef]
- Angellotti, M.C.; Bhuiyan, S.B.; Chen, G.; Wan, X. CodonO: Codon usage bias analysis within and across genomes. Nucleic Acids Res. 2007, 35, W132–W136. [Google Scholar] [CrossRef]
- Lavner, Y.; Kotlar, D. Codon bias as a factor in regulating expression via translation rate in the human genome. Gene 2005, 345, 127–138. [Google Scholar] [CrossRef]
- Tang, W.; Chen, Y. Ultrastructural observation on the chin adhesive apparatus and subgenus division of Pseudogastromyzon. Acta Zool. Sin. 1996, 42, 231–237. [Google Scholar]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2016, 45, e18. [Google Scholar] [CrossRef]
- Burland, T.G. DNASTAR’s Lasergene sequence analysis software. Methods Mol. Biol. 2000, 132, 71–91. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. The codon Adaptation Index—A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef]
- Ermolaeva, M.D. Synonymous codon usage in bacteria. Curr. Issues Mol. Biol. 2001, 3, 91–97. [Google Scholar] [PubMed]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Ikemura, T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985, 2, 13–34. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Vaidya, G.; Lohman, D.J.; Meier, R. SequenceMatrix: Concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Rabosky, D.L.; Chang, J.; Title, P.O.; Cowman, P.F.; Sallan, L.; Friedman, M.; Kaschner, K.; Garilao, C.; Near, T.J.; Coll, M.; et al. An inverse latitudinal gradient in speciation rate for marine fishes. Nature 2018, 559, 392–395. [Google Scholar] [CrossRef]
- Nakatani, M.; Miya, M.; Mabuchi, K.; Saitoh, K.; Nishida, M. Evolutionary history of Otophysi (Teleostei), a major clade of the modern freshwater fishes: Pangaean origin and Mesozoic radiation. BMC Evol. Biol. 2011, 11, 177. [Google Scholar] [CrossRef]
- Cui, L.; Dong, Y.; Liu, F.; Gao, X.; Zhang, H.; Li, L.; Cen, J.; Lu, S. The first two complete mitochondrial genomes for the family Triglidae and implications for the higher phylogeny of Scorpaeniformes. Sci. Rep. 2017, 7, 416–426. [Google Scholar] [CrossRef]
- Yu, F.; Yu, J.; Zhou, Y.; Yan, J.; Fang, Y.; Wang, W.J.; Yang, Z. Phylogenetic study of Ameiurus melas based on complete mitochondrial DNA sequence. Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 4706–4707. [Google Scholar] [CrossRef]
- Lee, S. Complete mitochondrial genome of the Korean flying fish Cheilopogon doederleinii (Beloniformes, Exocoetidae): Mitogenome characterization and phylogenetic analysis. Mitochondrial DNA Part B Resour. 2016, 1, 901–902. [Google Scholar] [CrossRef]
- Lee, J.S.; Miya, M.; Lee, Y.S.; Kim, C.G.; Park, E.H.; Aoki, Y.; Nishida, M. The complete DNA sequence of the mitochondrial genome of the selffertilizing fish Rivulus marmoratus (Cyprinodontiformes, Rivulidae) and the first description of duplication of a control region in fish. Gene 2001, 280, 1–7. [Google Scholar] [CrossRef]
- Shen, Z.; Sheng, Q.; Jin, Z.; Zhang, Y.; Lv, H. Mitogenome Characterization of a Vulnerable Gastromyzontid Fish, Beaufortia pingi (Teleostei: Gastromyzontidae): Genome Description and Phylogenetic Considerations. J. Ichthyol. 2023, 63, 735–746. [Google Scholar] [CrossRef]
- Oleinik, A.G.G.; Kukhlevsky, A.D.D.; Skurikhina, L.A.A.; Shubina, E.A.A. Complete mitochondrial genomes of Gritzenko’s charr Salvelinus gritzenkoi and Miyabe charr Salvelinus malma miyabei and their interpretation regarding intraspecific taxonomy. Mol. Biol. Rep. 2023, 50, 6273–6278. [Google Scholar] [CrossRef]
- Peng, Z.; Wang, J.; He, S. The complete mitochondrial genome of the helmet catfish Cranoglanis bouderius (Silurifonnes: Cranoglanididae) and the phylogeny of otophysan fishes. Gene 2006, 376, 290–297. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kweon, H.S.; Kim, I.C.; Lee, Y.M.; Kim, J.M.; Lee, J.S. The complete mitochondrial genome of the floating goby, Gymnogobius petschiliensis (Perciformes, Gobiidae). Mol. Cells 2004, 17, 446–453. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondrial. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Chung, H.H.; Kamar, C.K.A.; Lim, L.W.K.; Roja, J.S.R.; Liao, Y.S.; Lam, T.T.Y.; Chong, Y.L. Sequencing and characterization of complete mitogenome DNA of Rasbora tornieri (Cypriniformes: Cyprinidae: Rasbora) and its evolutionary significance. J. Genet. 2020, 99, 455–477. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, S.; Tian, Z.; Cheng, S.; Wang, D.; Xu, S. The complete mitochondrial genome of Chinese minnow (Rhynchocypris oxycephalus) and its phylogenetic analyses. Mitochondrial DNA Part B Resour. 2021, 6, 2177–2179. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Brown, W.M.; Boore, J.L. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc. Natl. Acad. Sci. USA 2000, 97, 13738–13742. [Google Scholar] [CrossRef]
- Lynch, M. Mutation accumulation in nuclear, organelle, and prokaryotic transfer RNA genes. Mol. Biol. Evol. 1997, 14, 914–925. [Google Scholar] [CrossRef]
- Cao, M.-L.; Nie, Y.; Fu, Y.-T.; Li, R.; Yi, X.-L.; Xiong, J.; Liu, G.-H. Characterization of the complete mitochondrial genomes of five hard ticks and phylogenetic implications. Parasitol. Res. 2023, 122, 1907–1913. [Google Scholar] [CrossRef]
- Zhang, X.; Bauman, N.; Brown, R.; Richardson, T.H.; Akella, S.; Hann, E.; Morey, R.; Smith, D.R. The mitochondrial and chloroplast genomes of the green alga Haematococcus are made up of nearly identical repetitive sequences. Curr. Biol. 2019, 29, R736–R737. [Google Scholar] [CrossRef]
- Carvalho, E.L.; Wallau, G.L.; Rangel, D.L.; Machado, L.C.; Pereira, A.B.; Victoria, F.D.C.; Boldo, J.T.; Pinto, P.M. Phylogenetic positioning of the Antarctic alga Prasiola crispa (Trebouxiophyceae) using organellar genomes and their structural analysis. J. Phycol. 2017, 53, 908–915. [Google Scholar] [CrossRef]
- Zhang, C.; Li, Q.; Meng, Q.; Wang, W.; Cheng, Y.; Wu, X. Sequence and phylogenetic analysis of the complete mitochondrial genome for Hepu mitten crab (Eriocheir hepuensis) from Nanjiujiang River basin. Mitochondrial DNA Part B Resour. 2019, 4, 3890–3891. [Google Scholar] [CrossRef]
- Mthethwa, S.; Bester-van der Merwe, A.E.; Roodt-Wilding, R. Addressing the complex phylogenetic relationship of the Gempylidae fishes using mitogenome data. Ecol. Evol. 2023, 13, e10217. [Google Scholar] [CrossRef]
- Zhang, Z.; Cheng, Q.; Ge, Y. The complete mitochondrial genome of Rhynchocypris oxycephalus (Teleostei: Cyprinidae) and its phylogenetic implications. Ecol. Evol. 2019, 9, 7819–7837. [Google Scholar] [CrossRef]
- Kartavtsev, Y.P.; Jung, S.O.; Lee, Y.M.; Byeon, H.K.; Lee, J.S. Complete mitochondrial genome of the bullhead torrent catfish, Liobagrus obesus (siluriformes, amblycipididae): Genome description and phylogenetic considerations inferred from the cyt b and 16s rRNA genes. Gene 2007, 396, 13–27. [Google Scholar] [CrossRef]
- Xu, W.; Lin, S.; Liu, H. Mitochondrial genomes of five Hyphessobrycon tetras and their phylogenetic implications. Ecol. Evol. 2021, 11, 12754–12764. [Google Scholar] [CrossRef]
- Lv, W.Q.; Jiang, H.F.; Bo, J.; Wang, C.; Yang, L.D.; He, S.P. Comparative mitochondrial genome analysis of Neodontobutis hainanensis and Perccottus glenii reveals conserved genome organization and phylogeny. Genomics 2020, 112, 3862–3870. [Google Scholar] [CrossRef]
- Zhao, J.L.; Wang, W.W.; Li, S.F.; Cai, W.Q. Structure of the Mitochondrial DNA Control Region of the Sinipercine Fishes and Their Phylogenetic Relationship. Acta Genet. Sin. 2006, 33, 793–799. [Google Scholar] [CrossRef]
- Lee, W.J.; Conroy, J.; Howell, W.H.; Kocher, T.D. Structure and evolution of teleost mitochondrial control regions. J. Mol. Evol. 1995, 41, 54–66. [Google Scholar] [CrossRef]
- Shadel, G.S.; Clayton, D.A. Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem. 1997, 66, 409–435. [Google Scholar] [CrossRef]
- Igawa, T.; Kurabayashi, A.; Usuki, C.; Fujii, T.; Sumida, M. Complete mitochondrial genomes of three neobatrachian anurans: A case study of divergence time estimation using different data and calibration settings. Gene 2008, 407, 116–129. [Google Scholar] [CrossRef]
- Zhang, K.; Zhu, K.; Liu, Y.; Zhang, H.; Gong, L.; Jiang, L.; Liu, L.; Lu, Z.; Liu, B. Novel gene rearrangement in the mitochondrial genome of Muraenesox cinereus and the phylogenetic relationship of Anguilliformes. Sci. Rep. 2021, 11, 2411. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhao, L.; Song, N.; Gao, T. The complete mitochondrial genome of Johnius grypotus (Perciformes: Sciaenidae). Mitochondrial DNA 2013, 24, 504–506. [Google Scholar] [CrossRef]
- Sharp, P.M.; Tuohy, T.M.F.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef]
- Bulmer, M. The selection-mutation-drift theory of synonymous codon usage. Genetics 1991, 129, 897–907. [Google Scholar] [CrossRef]
- Li, S. Study on the homalopterid fishes from Fujian province, China. J. Fish. China 1998, 22, 260–264. [Google Scholar]
- Wei, J.; Wu, R.; Xiao, Y.; Zhang, H.; Jawad, L.A.; Wang, Y.; Liu, J.; Al-Mukhtar, M.A. Validity of Pampus liuorum Liu & Li, 2013, Revealed by the DNA Barcoding of Pampus Fishes (Perciformes, Stromateidae). Diversity 2021, 13, 618. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Noncanonical Match Base Pairs | |||||
|---|---|---|---|---|---|---|
| A-A | A-C | C-C | C-U | G-U | U-U | |
| P. fasciatus jiulongjiangensis | 2 | 12 | 1 | 1 | 22 | 3 |
| P. fangi | 3 | 8 | 2 | 1 | 31 | 3 |
| P. fasciatus fasciatus | 2 | 11 | 1 | 1 | 24 | 3 |
| P. meihuashanensis | 2 | 12 | 1 | 1 | 24 | 3 |
| P. changtingensis changtingensis | 4 | 7 | 2 | − | 35 | 2 |
| P. changtingensis tungpeiensis | 3 | 8 | 2 | − | 36 | 3 |
| P. cheni | 2 | 11 | 2 | − | 26 | 3 |
| P. laticeps | 2 | 10 | 2 | − | 24 | 3 |
| P. lianjiangensis | 2 | 9 | 2 | − | 26 | 3 |
| P. myersi | 2 | 9 | 2 | − | 27 | 3 |
| P. peristictus | 2 | 12 | 2 | − | 26 | 3 |
| Species | Termination-Associated Sequence (TAS) | Central Conserved Domain (CCD) | Conserved Sequence Block (CSB) | ||||
|---|---|---|---|---|---|---|---|
| CSB-F | CSB-E | CSB-D | CSB-1 | CSB-2 | CSB-3 | ||
| P. fasciatus jiulongjiangensis | TACATCTATGTAATATCACCAA | ATGTAGTAAGAGACCACC | AGGGACAATAATCGTGGGGGT | TATTACTGGCATCTG | TTCATCATTAAAAGACATA | CAAACCCCCTTACCCCC | TGTCAAACCCCGAAACCA |
| P. fangi | TACATATATGTATTATCACCATT | ATGTAGTAAGAGACCACC | AGGGACAATAATCGTGGGGGT | TATTACTGGCATCTG | TTCATCATTAAAAGACATA | CAAACCCCCTTACCCCC | TGTCAAACCCCGAAACCA |
| P. fasciatus fasciatus | TACATCTATGTAATATCACCAA | ATGTAGTAAGAGACCACC | AGGGACAATAATTGTGGGGGT | TATTACTGGCATCTG | TTCATCATTAAAAGACATA | CAAACCCCCTTACCCCC | TGTCAAACCCCGAAACCA |
| P. meihuashanensis | TACATCTATGTAATATCACCAA | ATGTAGTAAGAGACCACC | AGGGACAATAATCGTGGGGGT | TATTACTGGCATCTG | TTCATCATTAAAAGACATA | CAAACCCCCTTACCCCC | TGTCAAACCCCGAAACCA |
| P. changtingensis changtingensis | TACATATATGTATTATCACCAT | ATGTAGTAAGAGACCACC | AGGGACAATAATCGTGGGGGT | TATTACTGGCATCTG | TTCATCATTAAAAGACATA | CAAACCCCCCTACCCCC | TGTCAAACCCCGAAACCA |
| P. changtingensis tungpeiensis | TACATATATGTATTATCACCAT | ATGTAGTAAGAGACCACC | AGGGACAATAATCGTGGGGGT | TATTACTGGCATCTG | TTCATCATTAAAAGACATA | CAAACCCCCTTACCCCC | TGTCAAACCCCGAAACCA |
| P. cheni | TACATATATGTAATATCACCAA | ATGTAGTAAGAGACCACC | AGGGACAATAATTGTGGGGGT | TATTACTGGCATCTG | TTCATCATTAAAAGACATA | CAAACCCCCTTACCCCC | TGTCAAACCCCGAAACCA |
| P. laticeps | TACATTCATGTAATATCACCAA | ATGTAGTAAGAGACCACC | AGGGACAATAATCGTGGGGGT | TATTACTGGCATCTG | TTCATCATTAAAAGACATA | CAAACCCCCTTACCCCC | TGTCAAACCCCGAAACCA |
| P. lianjiangensis | TACATTCATGTAATATCACCAA | ATGTAGTAAGAGACCACC | AGGGACAATAATCGTGGGGGT | TATTACTGGCATCTG | TTCATCATTAAAAGACATA | CAAACCCCCTTACCCCC | TGTCAAACCCCGAAACCA |
| P. myersi | TACATACATGTATTATCACCAA | ATGTAGTAAGAGACCACC | AGGGACAATAATCGTGGGGGT | TATTACTGGCATCTG | TTCATCATTAAAAGACATA | CAAACCCCCTTACCCCC | TGTCAAACCCCGAAACCA |
| P. peristictus | TACATATATGTAATATCACCAA | ATGTAGTAAGAGACCACC | AGGGACAATAATTGTGAGGGT | TATTACTGGCATCTG | TTCATCATTAAAAGACATA | CAAACCCCCTTACCCCC | TGTCAAACCCCGAAACCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Zhang, S.; Tian, Z.; Wang, Y.; Xu, S.; Wang, D. Comprehensive Analysis of Phylogenetic Relationship and Optimal Codons in Mitochondrial Genomes of the Genus Pseudogastromyzon. Animals 2024, 14, 495. https://doi.org/10.3390/ani14030495
Zhang C, Zhang S, Tian Z, Wang Y, Xu S, Wang D. Comprehensive Analysis of Phylogenetic Relationship and Optimal Codons in Mitochondrial Genomes of the Genus Pseudogastromyzon. Animals. 2024; 14(3):495. https://doi.org/10.3390/ani14030495
Chicago/Turabian StyleZhang, Cheng, Shun Zhang, Zhe Tian, Yajun Wang, Shanliang Xu, and Danli Wang. 2024. "Comprehensive Analysis of Phylogenetic Relationship and Optimal Codons in Mitochondrial Genomes of the Genus Pseudogastromyzon" Animals 14, no. 3: 495. https://doi.org/10.3390/ani14030495