
Abstract
The high biodiversity and strong population structure of freshwater fauna has often been attributed to historical geological and climatic alterations. The impact of these historical changes on obligate freshwater species on a small geographical scale has not been well understood due to the lack of fine-scale comparative phylogeographic studies. Strong population structure has been reported in a goby and a caridean shrimp in Hong Kong, a small but highly developed city in South China, but the common drivers of population differentiation in freshwater fauna in this region remain unclear. This study examined the fine-scale phylogeographic patterns of two freshwater loaches, Schistura fasciolata and Pseudogastromyzon myersi in Hong Kong, using sequence data of mitochondrial control region and two nuclear markers (interphotoreceptor retinoid binding protein gene 2 and ribosomal protein S13 gene). Results show that they exhibit pronounced population structure as supported by high and significant ΦST. Phylogenetic analyses based on the control region reveal six and three distinct lineages in S. fasciolata and P. myersi, respectively. Phylogeographic structure of both species generally follows the paleodrainage pattern, though P. myersi shows a shallower structure on the Mainland, perhaps due to their higher mobility. Most of these lineages diverged during the Pliocene and Late Pleistocene, a period with marked sea-level fluctuations. In a broader context, this suggests that sea-level fluctuation played an important role in shaping even the fine-scale population structure of freshwater fish in South China, implying that the genetic diversity of this fauna may be higher than expected.
Similar content being viewed by others


Introduction
Understanding the processes shaping the distribution and population structure of freshwater-restricted fauna has become a popular topic in biogeographic study with significant implications for conservation management. Since these organisms complete their entire life cycle in a freshwater environment, inter-drainage dispersal through the sea and over land is not possible (Burridge et al., 2008). However, many of these organisms are wide-spread, inhabiting drainages separated by land and marine barriers. Their broad distribution has often been attributed to significant re-arrangements in the drainage. For instance, river capture is believed to effectively influence the dispersal among populations of freshwater fish, Galaxias sp., in previously isolated drainages in New Zealand (Burridge et al., 2008). While historical river capture events are not frequently documented, sea level fluctuation and climate change have been subjected to intensive study and could also shape the demography and connectivity of freshwater fish populations. Temperature reduction during the Last Glacial Maximum in the Pleistocene epoch, for example, is argued to have been responsible for population bottlenecks in many freshwater fish populations (for example, Iberian Squalius, Almada and Sousa-Santos, 2010). Simultaneously, the sea level regression associated with Last Glacial Maximum exposed a confluence of drainages that are currently isolated by marine barriers, and thus allowed the dispersal of fish among otherwise isolated drainages (Swartz et al., 2007). While these geological and climatic changes could facilitate inter-drainage dispersal of freshwater fauna, their effects are often intermittent; hence fine-scale population genetic structure is expected to be prevalent in obligate freshwater species today (Dudgeon et al., 2006). To date, the impact of past geological and climatic events on inter-drainage dispersal has mostly been investigated in major river systems over a broad geographic scale (for example, Huey et al., 2013), and such studies have been very limited in East Asia. A phylogeographic study on the catfish Hemibagrus guttatus suggests the historical sea-level fluctuations might have shaped the genetic structure and led to fragmentation among its populations in the Pearl River (Yang and He, 2008). However, without sufficient geological evidence illustrated in this study, the timing and processes of diversification of H. guttatus in this area remain unclear. In-depth studies on the fine-scale phylogeographic pattern of freshwater fish would shed light on the evolutionary history in this region, which harbours very high biodiversity, much of which is unexplored and yet under imminent threat due to urbanisation and anthropogenic activities. If the drivers and processes of fine-scale population differentiation are found to be common and operate at a similar magnitude as on a larger geographical scale, it would imply that the extent of freshwater fauna diversity in South China may be much higher than expected.
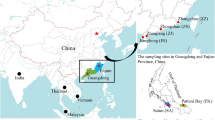
Hong Kong is a small territory (1140 km2) located in South China with a well-studied geological history. A high diversity of freshwater fish (more than 160 recorded species) can be found within a dense fluvial system, which presents an ideal system for fine-scale phylogeographic study aiming to assess the significance of paleo-climatic and geological events on inter-drainage dispersal of freshwater fish. The city consists of the ‘Mainland’ (Kowloon Peninsula and the New Territories), two large islands (Hong Kong Island and Lantau Island), and over 200 small islands (Figure 1). Most of the streams in Hong Kong, though closely located, have independent headwaters and form isolated drainage systems that are compact and short, with low stream order (Fyfe, 2000). As such, freshwater fauna is expected to exhibit low contemporary inter-drainage connectivity on a small geographic scale. Indeed, recent population genetic studies of freshwater shrimp, Caridina cantonensis, revealed strong genetic divergence and a low level of gene flow among populations within and among drainage systems (Tsang et al., 2016). The freshwater goby, Rhinogobius duospilus, which is a sedentary stream fish (Ho and Dudgeon, 2015), showed even stronger population differentiation and the pattern of population structure follows the Late Pleistocene paleodrainage pattern (Wu et al., 2016). However, with only two ecologically different and phylogenetically distant freshwater species examined in this region, it is unclear whether the observed patterns are common in multiple freshwater taxa in Hong Kong, and whether there were general historical causes that similarly shaped the population genetic structure of local stream inhabitants.

Current drainage pattern (blue) and late Pleistocene paleodrainage pattern (grey) of Hong Kong showing the sampling localities of S. fasciolata and P. myersi. Localities are marked as squares (for both species), circles (for S. fasciolata only) and triangles (for P. myersi only). Two major geographical barriers, Tai Mo Shan and Ma On Shan, are indicated as dotted lines representing the ridge of the two mountain ranges. A full color version of this figure is available at the Heredity journal online.
The unique geological settings and past sea-level changes may have played a significant role in shaping the population structure of freshwater fish, with some enhanced but some suppressed inter-drainage dispersal. The South China Sea level rose continuously from the early Miocene (23.03–15.97 mya) to the late Pliocene (3.60–2.58 mya) (Wang and Li, 2009; Ding et al., 2013; Zhou et al., 2015). Hence, the two main islands, Hong Kong and Lantau, were isolated from the Mainland for a prolonged period of time during a high sea level stand (Fyfe, 2000; Ding et al., 2013; Zhou et al., 2015), and so were their freshwater fauna on the islands from the Mainland counterpart. During the late Pleistocene (0.126–0.0177 mya), the sea level fluctuated rapidly with several major regressions recorded (Zong et al., 2009). The sea level in the South China Sea dropped from −80 to −130 m, and a large part of the continental shelf near the Pearl River mouth to the northern South China Sea was intermittently exposed during the late Quaternary glacial period (Eroglu et al., 2012). The two main islands thus were connected for only a short time to the Mainland, potentially permitting dispersal between the Mainland and the island populations. Furthermore, on the Mainland, the regressions allowed isolated drainages in Pat Sin Range and Ma On Shan to form a common confluence at the now submerged northeast basin (Tolo Harbour) before draining to the South China Sea (Figure 1). Therefore, the freshwater fish populations from the northeast possibly exhibit a higher level of historical gene flow than those from other regions of Hong Kong.
This study aims to understand the association between the evolutionary history of benthic freshwater fish and past climatic and geological changes in Hong Kong. The target species are two loaches, Schistura fasciolata and Pseudogastromyzon myersi. They are small benthic balitoroid loaches (TL<10 cm, Ho and Dudgeon, 2015) that widely coexist from middle to upper streams in Hong Kong and obligate to freshwater (Dudgeon, 1992). There are, however, subtle differences in their ecological traits: S. fasciolata is commonly found in East Asia streams on both stony and sandy substrates and mainly feeds on benthic micro-invertebrates and algae, while P. myersi is an algivorous loach endemic to the Guangdong province of mainland China that prefers streams with a stony substratum and abundant algal supply (Lee et al., 2004; Yang and Dudgeon, 2009). A recent mark-recapture study of these species in Hong Kong suggests that these species are highly sedentary, with a high recapture rate (62% of P. myersi and 67% of S. fasciolata) comparable to that of the freshwater goby R. duospilus (70%; Ho and Dudgeon, 2015). By analysing the phylogeographic patterns based on sequence variation in the mitochondrial control region (CR), the nuclear interphotoreceptor retinoid binding protein gene 2 (IRBP), and an exon-primed intron-crossing marker from ribosomal protein S13 (R13), we tested (1) if past climatic and geological events have exerted similar effects on the phylogeography of these sympatric species, and (2) if their highly sedentary nature confers a similar level of fine-scale genetic differentiation in these species.
Materials and methods
Specimens for S. fasciolata and P. myersi were collected from 33 sites in Hong Kong between 2013 and 2016 using hand nets and fish cages from 27 and 13 sampling localities, respectively (Figure 1). Sample sizes were not uniform due to the difficulties in obtaining specimens in some localities with a small population size. All collected individuals were>1.5 cm (total length). Specimens were stored in an icebox during transportation then killed by freezing and preserved in 95% ethanol before DNA extraction.
Total genomic DNA was extracted from skeletal muscle or fin clip from one side of the body using QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. The primer pairs used for PCR were ThrF and 12SR (Miya and Nishida, 1999; Cheng et al., 2012) for the complete CR sequence, IRBP109F and IRBP1001R for the IRBP fragment (Chen et al., 2008), and 36298E1_F and 36298E1_R for R13 fragment (Li et al., 2010). PCR amplification of CR was performed in a 40 μl reaction volume containing 27.2 μl sterile distilled water, 4 μl of 10 × PCR reaction buffer, 3.2 μl of dNTP (2.5 mM each), 2.0 μl each of primers (10 μM), 0.2 μl Taq polymerase and 2 μl DNA template. The thermal profile was as follows: initial denaturation at 95 °C for 30 s followed by 34 cycles of denaturation for 30 s, annealing at 55 °C for 30 s and extension at 72 °C for 1.5 min, and a final extension at 72 °C for 5 min. The PCR mixture and profile for the two nuclear markers followed the same procedures as CR amplification except 4 μl DNA template were added to the reaction mix and annealing temperature was set at 48 °C for the thermal cycling conditions. PCR products were visualised using electrophoresis in 1.0% agarose gel using 0.5 × Tris/Borate/EDTA (TBE) buffer and purified using the Millipore PCR plate (Merck Millipore, Billerica, USA) for products with a clear single band. Where there were multiple bands present, the desired DNA band was cut and eluted using the QIAquick PCR purification Kit (Qiagen). Sequencing was performed using the same set of primers as in PCR amplification on an Applied Biosystems (ABI) (Foster City, CA, USA) 3710xl automated sequencer, following the standard cycle sequencing protocol with the ABI Big-dye Ready-Reaction mix kit.
Sequences were manually edited by using Seqtrace v0.9.0 (Stucky, 2012) followed by alignment using MUSCLE implemented in MEGA v7.0.18 (Kumar et al., 2016). Alleles with heterozygous SNPs for the two nuclear markers were inferred by PHASE v2.1 (Stephens and Scheet, 2005) implemented in DnaSP v5.0 (Librado and Rozas, 2009) using default settings. Population genetic diversity indices, including haplotype diversity (h), nucleotide diversity (π), Tajima’s D and Fu’s FS, were calculated for each site in each species using DnaSP v5.0. Neutrality tests (Tajima’s D and Fu’s FS) were conducted to infer the demographic history of each species to determine if the mutations were selectively neutral. Demographic expansion events were also examined by calculating Harpending’s raggedness index (r) and mismatch distribution analysis performed using Arlequin v3.5.2.2 (Excoffier and Lischer, 2010). The pairwise genetic distance was calculated with the Taimura–Nei model using the ape package in R v.3.2.2 (Paradis et al., 2004). To reveal the genealogical relationships among haplotypes, haplotype networks at 95% confidence level were constructed for all markers using TCS v.1.13 (Clement et al., 2000). The maximum connection unit was set to 40 in order to connect distantly related haplotype groups. Haplotype networks were visualised using Gephi v0.8.2 (Bastian et al., 2009).
We used SAMOVA v2.0 (Dupanloup et al., 2002) to delineate the best clustering of population-groups in terms of genetic distinctiveness in the three markers. The default setting specified for DNA sequence was used, with T92 model as the molecular substitution model and an initial configuration of groups at 100. The optimal number of groups (K) was selected based on the highest ΦCT value and configuration with no single population-group included (except Lantau Island populations, see Results). Analysis of Molecular Variance (AMOVA) and pairwise genetic differentiation values (ΦST) were then calculated for the partitions defined by SAMOVA v2.0.
Aligned CR sequences of both species were subjected to phylogenetic analysis and molecular dating. Phylogenetic analysis was not performed on nuclear markers because of the low number of informative sites for both species. The most appropriate model of nucleotide substitution was selected by JModelTest v2.1.7 (Posada, 2008) using the Akaike Information Criterion. The optimum model of substitution was HKY+I for the CR gene in both species. The CR sequence of S. thai (GenBank accession no.: DQ105264) was used as an outgroup in the phylogenetic analyses of S. fasciolata, while Pseudogastromyzon jiulongjiangensis (DQ105293) was used as an outgroup in the analysis of P. myersi. In the absence of fossil records and direct molecular clock calibration for the CR of S. fasciolata and P. myersi, we set the CR substitution rate, with reference to the mitochondrial cytochrome b (cyt b) clocks reported from their sister family Cobitidae (Liu et al., 2012), since cyt b and CR are linked during inheritance and have similar proportions of variable sites. Two comparable molecular clocks of cyt b were reported: 0.42% per lineage per myr for Cobitis taenia (Perdices and Doadrio, 2001) and 0.34% per lineage per myr for the genus Cobitis (Doadrio and Perdices, 2005). The relative mutation rate of the two genes, cyt b and CR, have been reported to be genus specific (Ruokonen and Kvist, 2002), and in the superfamily Cobitoidae, the mutation rate of CR is slower than that of cyt b (CR/ cyt b=0.78) in closely related species (Tang et al., 2006). By considering the reported clocks and ratio of mutation rate of CR and cyt b, two strict clocks of 0.25 and of 0.4% per lineage per myr, respectively, were applied in this study. The molecular clock hypothesis was not rejected by the maximum likelihood test which was implemented in MEGA v.7.0.18 (Kumar et al., 2016) and the distance based likelihood test implemented in DAMBE v.6.4.29 (Xia X, 2013) in both species. The phylogenetic analysis and estimation of divergence times were performed using *BEAST (Heled and Drummond, 2009) implemented in BEAST v1.8.2 (Drummond et al., 2012). Three independent analyses were performed with 2 000 000 steps, sampled every 2000 steps for each of the two rates in both species. The analysis was then verified to reach convergence with the chains mixing properly using Tracer v1.60. Replicated results were combined using LogCombiner v1.8.2. The maximum clade credibility trees with 25% burn-in were summarised in TreeAnnotator v1.8.2. The results of the phylogenetic analyses were then viewed with FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/). Maximum likelihood tree reconstruction was computed using MEGA v. 7.0.18 (Kumar et al., 2016) with settings of HKY+I model and 1000 bootstrap replicates.
Results
Schistura fasciolata
Aligned sequences of CR (853 bp) were obtained from 395 S. fasciolata individuals. Over the whole data set, there were 75 polymorphic sites and 35 haplotypes (Table 1). The genetic diversity of CR in S. fasciolata was high with an overall haplotype diversity (h) of 0.886. Haplotype diversity varied among localities, ranging from 0 to 0.861 (Table 1). The average nucleotide diversity (π) was 0.014. The highest nucleotide diversity was found on Lantau Island (ITF) (π=0.018), while some localities showed very low or no nucleotide diversity. Private haplotypes were observed in most of the localities. Sequences of IRBP (767 bp) and R13 (512 bp) were obtained from 291 and 189 individuals and the number of alleles were 18 and 40, respectively (Supplementary Table 1). The overall haplotype diversity (h) of the two nuclear markers were high (IRBP: 0.809; R13: 0.946), and exhibit differences in the degree of nucleotide diversity (π); the overall nucleotide diversity of IRBP was low (π=0.0037) while that of R13 was comparatively high (π=0.0194). These nuclear markers were not used in phylogenetic analysis because of the low number of informative sites.
Phylogenetic analysis and estimation of divergence time
The phylogenetic tree constructed from S. fasciolata CR haplotypes (Figure 2a) showed six monophyletic groups with strong statistical support (maximum likelihood bootstrap>80% and BI posterior probability>0.95) (Figure 3a): (1) the Pok Fu Lam lineage, which only occurred in the western part of Hong Kong Island, (2) the Central lineage, which was mostly found in the central part of the Mainland with a few sampled from the north-eastern part of the Mainland (NBP, NSH and NTK) and in the central region of Hong Kong Island, (3) the East lineage, which was found in the eastern part of the Mainland and Hong Kong Island, with a few from the north-eastern part of the Mainland (NSPT, NSH, NPS and NTK), (4) the North lineage, which was located at the north-eastern part of the Mainland (except NSPT), (5) the Lantau lineage, which was found on the Lantau Island, and (6) the Tong Fuk lineage, which contained only two individuals from site ITF in southern Lantau Island. Among these six lineages, the East lineage exhibited the widest distribution from the northern-most (NSPT) to the southern-most site (HSO). Most of the lineages exhibited allopatric distribution, while the Central, East and North lineages coexisted in two sites in the northeast area of the Mainland (NSH and NTK).

Bayesian inference tree of (a) S. fasciolata and (b) P. myersi based on mitochondrial control region sequences. Values above the branches represent bootstrap support from maximum likelihood (before slash) and Bayesian posterior probabilities (after slash). The geographic distribution of the terminal nodes (haplotypes) is indicated using the same colours as in the TCS networks and maps (see Figure 3). OG, Oligocene; MI, Miocene; PO, Pliocene; PS, Pleistocene. A full color version of this figure is available at the Heredity journal online.

Geographic distribution of haplotypes and TCS networks of (a,b) mitochondrial control region, (c,d) nuclear interphotoreceptor retinoid binding protein gene 2 and (e,f) ribosomal protein R13 of S. fasciolata. The number on each branch indicates the number of mutational steps between the haplotypes. The size of circle is proportional to the number of individuals with the particular haplotype. Current drainage pattern (blue lines) and paleodrainage pattern in the late Pleistocene (grey lines) are shown. The four paleodrainage basins are coloured (yellow: Southwest basin; green: Middle basin; blue: East basin; red: Northeast basin). A full color version of this figure is available at the Heredity journal online.
Based on BEAST estimation, the divergence of the six S. fasciolata lineages occurred from the late Miocene to the early Pleistocene epoch (Figure 2a, see also Supplementary Table 7). The Tong Fuk lineage diverged from the ancestral population from 10.12 (6.70–14.04, 95% HPD; 0.25% clock) to 6.32 (4.24–8.78, 95% HPD; 0.4% clock) mya during the late Miocene. The others then split into two clades (Pok Fu Lam+Central and East+North+Lantau) from 5.61 (3.66–7.86, 95% HPD) to 3.51 (2.30–4.96, 95% HPD) mya during the Late Miocene to Early Pliocene, followed by the divergence between Lantau lineage and East+North populations from 3.46 (2.12–5.16, 95% HPD) to 2.17 (1.29–3.20, 95% HPD) mya in the late Pliocene. From there on, the Pok Fu Lam lineage and the Central lineage split at approximately 1.97 (1.05–3.11, 95% HPD) to 1.25 (0.68–1.97, 95% HPD) mya and the East lineage and North lineage split at approximately 1.82 (1.04–2.81, 95% HPD) to 1.14 (0.64–1.75, 95% HPD) mya.
The phylogenetic relationship among populations could not be resolved using nuclear markers, and hence, were excluded from the phylogenetic study in both species. This is because the statistical support of nearly all nodes from trees based on these markers were too low, probably due to the low resolution of nuclear markers at the population level.
Demographic and population differentiation analysis
The haplotype networks of S. fasciolata showed a similar partition to the phylogenetic tree. The CR haplotype network (Figure 3b) was composed of four major haplotype groups which corresponded to Pok Fu Lam+Central, East+North, Lantau and Tong Fuk lineages of the BI tree. A single, very distantly related Tong Fuk haplotype found in ITF was 35 mutation steps from the Lantau haplotype group. Haplotypes of Pok Fu Lam and Central lineages displayed a star shape, often interpreted as a sign of recent population expansion. However, the Tajima’s D and Fu’s Fs values of all but one (Tajima’s D of site NTY) were statistically insignificant. The networks of two nuclear markers showed a similar pattern to the CR network. In the IRBP network (Figure 3d), all haplotypes differed from their neighbouring ones by only one mutation step. Notably, the two Lantau Island sites each harboured a distinct haplotype. The R13 network revealed a finer division of haplotype groups than the IRBP network (Figure 3f). Similarly, the two sites in Lantau Island each had their specific haplotypes, which were 17–20 mutation steps apart.
SAMOVA defined seven population-groups in CR of S. fasciolata. Differing from the six lineages from BI tree, the populations in Pok Fu Lam and Tong Fuk were grouped with those on Hong Kong Island and Lantau, respectively, while the North lineage was subdivided into two population-groups. In addition, Lung Mei, a locality belonging to the East lineage in the BI tree was defined as a unique population. Pairwise, ΦST analyses of three loci revealed high and significant genetic differentiation across all population-groups (Table 2). Most of the comparisons between different localities were also high (> 0.4 in CR,>0.63 in R13 and>0.34 in IRBP) and significant (P<0.01) (Supplementary Tables 2 and 3). Global ΦCT values were high and significant for all three loci (Supplementary Table 8).
Pseudogastromyzon myersi
Aligned sequences of CR (950 bp) were obtained from 210 P. myersi individuals (Table 3). In total, there were 41 polymorphic sites and 13 haplotypes. The genetic diversity of CR was high, with an average haplotype diversity of 0.816. In contrast, the nucleotide diversity was relatively low in most localities, ranging from zero (NNT, NSPA, NTP and MMB) to 0.007 (MSD). The sequences of IRBP (762 bp) and R13 (478 bp) were obtained from 108 and 111 individuals, respectively. Only six and five haplotypes were detected in IRBP and R13, respectively (Supplementary Table 4). The genetic diversity of both nuclear markers was low, with a haplotype diversity of 0.409 and 0.260, and a nucleotide diversity of 0.0007 and 0.0038, respectively.
Phylogenetic analysis and estimation of divergence time
The phylogenetic tree constructed from P. myersi CR haplotypes revealed three lineages with moderate statistical support (maximum likelihood bootstrap >75% and BI posterior probability>0.90 Figure 2b). The three groups exhibited a distinct distribution in Hong Kong and are referred to as the Mainland, Tai Mo Shan and Lantau lineages hereafter. The Mainland lineage was found in all localities on the Mainland except for site NCL, southwest of Tai Mo Shan (Figure 4a). The Tai Mo Shan lineage was only found in the two localities (NCL and MSD) at the southwest of Tai Mo Shan. Site IWL on Lantau Island was the only locality where the Lantau lineage was found. All individuals in each locality belonged to the same lineage except for MSD, where the Mainland and Tai Mo Shan lineages coexisted.

Geographic distribution of haplotypes and TCS networks of (a,b) mitochondrial control region, (c,d) nuclear interphotoreceptor retinoid binding protein gene 2 and (e,f) ribosomal protein R13 of P. myersi. The number on each branch indicates the number of mutational steps between the haplotypes. The size of each circle is proportional to the number of individuals with the particular haplotype. Current drainage pattern (blue lines) and paleodrainage pattern in the late Pleistocene (grey lines) are shown. The four paleodrainage basins are coloured (yellow: Southwest basin; green: Middle basin; blue: East basin; red: Northeast basin). A full color version of this figure is available at the Heredity journal online.
We estimated an early Pliocene to early Pleistocene divergence of the three P. myersi lineages (Figure 2b, see also Supplementary Table 7). The Mainland lineage first diverged from the others at approximately 5.75 (3.57–8.40, 95% HPD; 0.25% clock) to 3.61 mya (2.27–5.31, 95% HPD; 0.4% clock) during the late Miocene to early Pliocene. The Tai Mo Shan lineage and Lantau lineage diverged from 3.69 (1.97–5.87, 95% HPD) to 2.32 (1.20–3.66, 95% HPD) mya in the late Pliocene.
Demographic and population differentiation analysis
The Tajima’s D and Fu’s Fs values of all localities were not statistically significant. The CR haplotype network revealed three groups equivalent to the three lineages defined by the BI tree. The network of the two nuclear markers (IRBP and R13) showed a similar pattern and grouping to the CR network (Figures 4d and f). Yet, the numbers of mutation steps between haplotype groups were lower than those in the CR network due to the more conservative nature of the nuclear markers (see Supplementary Table 9). Both CR and nuclear marker networks reveal pronounced population genetic structure in this species.
SAMOVA defined three population-groups of P. myersi which are equivalent to the three lineages in BI tree in all three loci. Pairwise ΦST analyses of the three loci reveal a high level of genetic differentiation across all population-groups; all the values between the three groups were high (> 0.7) and significant (P<0.01) (Table 4). Most of the values between different localities within groups were high (> 0.7) and significant (P<0.01) in the CR marker (Supplementary Tables 5 and 6). A high global ΦCT value of 0.83 (P<0.01) was calculated from AMOVA analyses on CR (Supplementary Table 8). The global ΦCT values for the two nuclear markers were higher than 0.90 (P<0.01). The genetic variations among groups were much larger than the variations within groups or within localities.
Discussion
Phylogeographic inference
We present herein the first phylogeographic study of balitorid loaches on a small geographical scale in Hong Kong, which reveals pronounced population structure in both studied species. Six and three deeply divergent lineages are recovered in S. fasciolata and P. myersi, respectively. A high level of differentiation is not only observed among lineages, but also between adjacent localities, suggesting limited gene flow between neighbouring streams separated by as little as 10 km. The high ΦST values are in congruence with results from previous studies on two other freshwater organisms in Hong Kong, which show high population differentiation between individual streams (Tsang et al., 2016; Wu et al., 2016). Moreover, by comparing and contrasting the phylogeographic structure of the two species studied, our study infers that climatic changes, sea level fluctuation and biological traits of organisms all play important roles in shaping the evolutionary history of loaches, on even a small geographical scale.
Our study reveals very similar temporal patterns and generally congruous spatial patterns of lineage divergence for the two loach species, in Hong Kong. Temporally, most of the lineages are estimated to have diverged during the late Miocene to the late Pliocene, which coincides with the marked climatic change in the late Miocene. East China had generally been humid from the Late Oligocene, until significant aridity developed in East Asia in the mid-Miocene and intensified during the late Miocene (Liu et al., 2009). Drought reduces hydrological connectivity among streams and is a major cause of freshwater habitat fragmentation (Covich et al., 2003). The prolonged aridity would have been able to significantly limit the headwater connectivity and reduce opportunities of flooding in the lower stream reaches, and thus facilitate lineage diversification of the two loach species in Hong Kong. This proposition is supported by other studies that suggested that low flow conditions weakened connectivity in freshwater fish species (for example, Etheostoma cragini, Labbe and Rausch, 2000). As there were no significant tectonic events recorded in this period, diversification due to drainage rearrangement caused by major geological changes is deemed less likely.
Spatially, our study shows that the degree of isolation among lineages on the major islands (Hong Kong Island and Lantau Island) and the Mainland is closely associated with the paleodrainage pattern and the location of common confluences. Both Lantau Island and Hong Kong Island are located very close to the Mainland, but the populations on the two islands show a distinct level of divergence with respect to the Mainland populations. The Lantau Island lineages diverged during the late Pliocene to the early Pleistocene in both species, coinciding with the rapid rise in sea level of the South China Sea during the late Pliocene (Wang and Li, 2009; Ding et al., 2013). The high sea level stand that separated the Mainland and Lantau Island might have acted as a strong geographic barrier to gene flow for the loaches. During the late Quaternary glacial cycle, the sea level fluctuated extensively, and a large part of the northern South China Sea was repeatedly exposed when the sea level was –120 m (Wang and Li, 2009). As a result, the Ma Wan Channel that separates Lantau Island and the Mainland would have been exposed and acted as a land bridge between the two. The paleodrainage in the western Mainland and Lantau Island converged far away at the Pearl River estuary which is situated around 15 km west of Lantau Island and the Mainland (Fyfe, 2000). This temporary connection between Lantau Island and the Mainland may have been temporally too short or spatially too distant for effective gene flow among freshwater fish populations on the two landmasses, especially for the loaches that inhabit the mid-stream and upper stream. By contrast, the confluence of Hong Kong Island and the Mainland palaeodrainage was close (< 1 km) to the Hong Kong Island, whereby population connectivity during periodical Quaternary low sea levels were possible. This might explain why S. fasciolata populations on Hong Kong Island share some haplotypes with those in the Mainland. Hence, the populations on Hong Kong Island were probably separated from those in the Mainland very recently by the last Holocene transgression. The effect of exposure of palaeodrainage confluence in facilitating gene flow is also evidenced in the mixing of S. fasciolata Central and East lineages in the northeast Mainland: the middle and east paleodrainages should have converged at the northeast lowland (that is, the current location of Tolo Habour) close to the shoreline (Fyfe, 2000).
Our results show that the evolution of the paleodrainage system might have been critical in shaping the population structure in S. fasciolata, suggested by the dissimilar haplotypes found on the two main islands (Figures 2 and 4a). This is in agreement with the findings of a recent phylogeographic study on a freshwater goby, R. duospilus in Hong Kong (Wu et al., 2016). Unfortunately, we were unable to sample P. myersi on Hong Kong Island in this study for comparison. More sampling of populations on Hong Kong Island is needed to confirm our hypothesis. Nonetheless, our results suggest that divergence of freshwater fish may also occur among islands that are extremely close to the coast. This implies high genetic diversity of aquatic fauna in the myriad islands along the southeastern coast of China. The population structure of other freshwater fish probably also follows the paleodrainage pattern to some extent, with the degree of similarity varying with species-specific life history traits.
Both extrinsic (for example, climatic and geological history) and intrinsic (for example, ecological specialisation, dispersal and reproductive behaviours) factors influence the population dispersal processes (Tibbets and Dowling, 1996). Many studies have reported the effects of intrinsic factors on evolutionary processes of closely related species and demonstrated the importance of species-specific ecological characteristics on dispersal (Koblmuller et al., 2007). Our analyses reveal a subtle difference in the degree of population structure between the two loach species. Generally, P. myersi exhibits a shallower genetic structure than S. fasciolata, most obviously among the Mainland populations. The difference might be attributed to their specific biological traits, particularly their mobility. S. fasciolata and P. myersi are well adapted to fast-flowing streams, but they may be swept downstream during a downpour in the rainy season. A recent mark-recapture study in Hong Kong reveals a marked difference in their mobility: rheophilic behaviour is common in balitoroid loaches, and P. myersi shows a particularly strong tendency to move upstream (Ho and Dudgeon, 2015). In contrast, S. fasciola is sedentary with limited movement along the stream. In addition, we observed gregarious behaviour in S. fasciolata in many localities which may restrict their mobility. It would be less likely for S. fasciolata swept down to lower stream reaches to migrate back to their original habitats than P. myersi. Therefore, we believe that the difference in mobility is a major factor leading to the more pronounced population structure in S. fasciolata.
We also observed a difference in the location of the major genetic break among the Mainland populations in the two loach species. The population subdivision of S. fasciolata is congruent with the late Pleistocene palaeodrainage pattern, which roughly comprises four drainages flowing west, central, east and northeast, with the Mainland populations primarily separated into two groups by the Ma On Shan ridge. Our results show that the populations located on the two sides of the ridge are genetically distinct from each other and do not share common haplotypes. This east–west separation is also observed on Hong Kong Island. However, for P. myersi, analyses of both mitochondrial and nuclear markers reveal that Tai Mo Shan ridge, instead of the Ma On Shan ridge is the major genetic break. This discrepancy in population structure between the two species remains to be explored.
Conservation implications
Located in the Indo-Burma biodiversity hotspot, Hong Kong has a high diversity of freshwater fauna, which are, unfortunately, severely threatened by rapid urban development. Although S. fasciolata and P. myersi inhabit upstream and mid-stream areas that are relatively distant from the highly developed coastal area, several habitats with populations of high levels of genetic differentiation are under serious threat of imminent urbanisation. The populations in Lantau Island (IWL and ITF) are under threat due to a proposed land development plan that aims to change the island into a major recreation and tourism zone. In addition, P. myersi warrants more management attention since it is uncommon and geographically more restricted. Furthermore, this species was historically found in both Guangdong and Hong Kong, but not recorded in a recent comprehensive survey in Guangdong (Zhao and Cui, 2011), probably due to habitat destruction there. In this study, we were only able to sample 13 P. myersi populations, most of which are found in the north Tai Mo Shan drainage. A more detailed survey is urgently needed to assess the extinction risk of P. myersi.
Conclusion
This study reveals marked population structure and similar temporal-spatial patterns of lineage divergence in two stream loaches, S. fasciolata and P. myersi, and provides insights into the evolutionary history of freshwater fish in South China. By comparing the phylogeographical pattern of the two species, we suggest that two historical events, namely the climatic change during the late Miocene and the major sea-level transgression during the Pliocene, played important roles in shaping the evolutionary history of freshwater fish in Hong Kong. The subtle discordance in their population structure is mainly attributed to their difference in mobility. The finding that considerable population structure exists on such a fine scale across multiple species implies that the overall freshwater biodiversity is higher than expected and warrants more investigation. Considering the current geographic isolation and strong genetic differentiation of Lantau Island populations of S. fasciolata and P. myersi, we identified them as populations with a high priority in biodiversity conservation, especially for P. myersi which has a restricted distribution.
Data archiving
Sequence data have been submitted to GenBank: accession numbers KY421988 - KY423282.
Accession codes
References
Almada V, Sousa-Santo C . (2010). Comparisons of the genetic structure of Squalius populations (Pisces, Cyprinidae) from rivers with contrasting histories, drainage areas and climatic conditions. Mol Biol Evol 57: 924–931.
Bastian M, Heymann S, Jacomy M . (2009). Gephi: an open source software for exploring and manipulating networks. International AAAI Conference on Weblogs and Social Media. North America. March 2009. Available at http://www.aaai.org/ocs/index.php/ICWSM/09/paper/view/154.
Burridge CP, Craw D, Fletcher D, Waters JM . (2008). Geological dates and molecular rates: fish DNA sheds light on time dependency. Mol Biol Evol 25: 624–633.
Chen WJ, Miya M, Saitoh K, Mayden RL . (2008). Phylogenetic utility of two existing and four novel nuclear gene loci in reconstructing Tree of Life of ray-finned fishes: the order Cypriniformes (Ostariophysi) as a case study. Gene 423: 125–134.
Cheng YZ, Xu TJ, Jin XX, Tang D, Wei T, Sun YY et al. (2012). Universal primers for amplification of the complete mitochondrial control region in marine fish species. Mol Biol 46: 727–730.
Clement M, Posada D, Crandall K . (2000). TCS: a computer program to estimate gene genealogies. Mol Ecol 9: 1657–1660.
Covich AP, Crowl TA, Scatena FN . (2003). Effects of extreme low flows on freshwater shrimps in a perennial tropical stream. Freshw Biol 48: 1199–1206.
Ding W, Li J, Li J, Fang Y, Yong T . (2013). Morphotectonics and evolutionary controls on the Pearl River Canyon system, South China Sea. Mar Geophys Res 34: 221–238.
Drummond AJ, Suchard MA, Xie D, Rambaut A . (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29: 1969–1973.
Doadrio I, Perdices A . (2005). Phylogenetic relationships among the Ibero-African cobitids (Cobitis, Cobitidae) based on cytochrome b sequence data. Mol Phylogenet Evol 37: 484–493.
Dudgeon D . (1992) Patterns and processes in stream ecology: A synoptic review of Hong Kong running waters. Schweizerbart: Stuttgart.
Dudgeon D, Arthington AH, Gessner MO, Kawabata ZI, Knowler DJ, Lévêque C et al. (2006). Freshwater biodiversity: importance, threats, status and conservation challenges. Biol Rev Camb Philos Soc 81: 163–182.
Dupanloup I, Schneider S, Excoffier L . (2002). A simulated annealing approach to define the genetic structure of populations. Mol Ecol 11: 2571–2581.
Eroglu S, Toprak S, Urgan O, Onur Ozge E., Denizbasi Arzu, Akoglu Haldun et al. (2012) The South China Sea: Paleoceanography and Sedimentology. Springer: The Netherlands.
Excoffier L, Lischer HEL . (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10: 564–567.
Fyfe JA . (2000) The Quaternary geology of Hong Kong. Hong Kong Geological Survey: Hong Kong.
Heled J, Drummond AJ . (2009). Bayesian inference of species trees from multilocus data. Molecular Biology and Evolution 27: 570–580.
Ho BSK, Dudgeon D . (2015). Movement of three stream-resident balitoroid loaches and a goby in a Hong Kong hillstream. Ecol Freshw Fish 25: 622–630.
Huey JA, Espinoza T, Hughes JM . (2013). Natural and anthropogenic drivers of genetic structure and low genetic variation in the endangered freshwater cod, Maccullochella mariensis. Conserv Genet 14: 997–1008.
Koblmuller S, Sefc KM, Duftner N, Warum M, Sturmbauer C . (2007). Genetic population structure as indirect measure of dispersalability in a Lake Tanganyika cichlid. Genetica 130: 121–131.
Kumar S, Stecher G, Tamura K . (2016). MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol 33: 1870–1874.
Labbe TR, Rausch KD . (2000). Dynamic of intermittent stream habitat regulate persistence of a threatened fish at multiple scales. Ecol Appl 10: 1774–1791.
Lee LF, Lam KS, Ng KY, Chan KT, Young LC . (2004) Field Guide to the Freshwater Fish of Hong Kong. Friends of the Country Parks and Cosmos Books Ltd: Hong Kong.
Yang L, He S . (2008). Phylogeography of the freshwater catfish Hemibagrus guttatus (Siluriformes, Bagridae): implications for South China biogeographyand influence of sea-level changes. Mol Phylogenet Evol 49: 393–398.
Li C, Riethoven JJ, Ma L . (2010). Exon-primed intron-crossing (EPIC) markers for non-model teleost fishes. BMC Evol Biol 10: 90.
Librado P, Rozas J . (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Liu L, Eronen JT, Fortelius M . (2009). Significant mid-latitude aridity in the middle Miocene of East Asia. Palaeogeography, Palaeoclimatology, Palaeoecology 279: 201–206.
Liu SQ, Mayden RL, Zhang JB, Yu D, Tang QY, Deng X et al. (2012). Phylogenetic relationships of the Cobitoidea (Teleostei: Cypriniformes) inferred from mitochondrial and nuclear genes with analyses of gene evolution. Gene 508: 60–72.
Miya M, Nishida M . (1999). Organization of the mitochondrial genome of a deep-sea fish, Gonostoma gracile (Teleostei: Stomiiformes): First example of transfer RNA gene rearrangements in bony fishes. Mar Biotechnol 1: 416–426.
Paradis E, Claude J, Strimmer K . (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20: 289–290.
Perdices A, Doadrio I . (2001). The molecular systematics and biogeography of the European cobitids based on mitochondrial DNA sequences. Mol Phylogenet Evol 19: 468–478.
Posada D . (2008). jModelTest: phylogenetic model averaging. Mol Biol Evol 25: 1253–1256.
Ruokonen M, Kvist L . (2002). Structure and evolution of the avian mitochondrial control region. Molecular Phylogenetics and Evolution 23: 422–432.
Stephens M, Scheet P . (2005). Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am J Hum Genet 76: 449–462.
Stucky BJ . (2012). SeqTrace: a graphical tool for rapidly processing DNA sequencing chromatograms. J Biomol Tech 23: 90–93.
Swartz ER, Skelton PH, Bloomer P . (2007). Sea-level changes, river capture and the evolution of populations of the Eastern Cape and fiery redfins (Pseudobarbus afer and Pseudobarbus phlegethon, Cyprinidae) across multiple river systems in South Africa. J Biogeogr 34: 2086–2099.
Tang Q, Liu H, Mayden R, Xiong B . (2006). Comparison of evolutionary rates in the mitochondrial DNA cytochrome b gene and control region and their implications for phylogeny of the Cobitoidea (Teleostei: Cypriniformes). Mol Phylogenet Evol 39: 347–357.
Tibbets CA, Dowling TE . (1996). Effects of intrinsic and extrinsic factors on population fragmentation in three species of North American minnows (Teleostei: Cyprinidae). Evolution 50: 1280–1292.
Tsang LM, Tsoi KH, Chan SKF, Chan TKT, Chu KH . (2016). Strong genetic differentiation among populations of the freshwater shrimp Caridina cantonensis in Hong Kong: implications for conservation of freshwater fauna in urban areas. Mar Freshwater Res 68: 187–194.
Wang P, Li Q . (2009) The South China Sea: Paleoceanography and sedimentology. Springer: Dordrecht.
Wu TH, Tsang LM, Chen IS, Chu KH . (2016). Multilocus approach reveals cryptic lineages in the goby Rhinogobius duospilus in Hong Kong streams: role of paleodrainage systems in shaping marked population differentiation in a city. Mol Phylogenet Evol 104: 112–122.
Xia X . (2013). DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution. Mol Biol Evol 30: 1720–1728.
Yang GY, Dudgeon D . (2009). Seasonal and inter-stream variations in the population dynamics, growth and secondary production of an algivorous fish (Pseudogastromyzon myersi: Balitoridae) in monsoonal Hong Kong. Freshw Biol 54: 1960–1976.
Zhao H, Cui K . (2011) Pseudogastromyzon myersi. The IUCN Red List of Threatened Species. e.T166199A6189602. http://dx.doi.org/10.2305/IUCN.UK.2011-2.RLTS.T166199A6189602.en (accessed 8 April 2016).
Zhou W, Gao X, Wang Y, Zhuo H, Zhu W, Xu Q et al. (2015). Seismic geomorphology and lithology of the early Miocene Pearl River Deepwater Fan System in the Pearl River Mouth Basin, northern South China Sea. Mar Pet Geol 68: 449–469.
Zong Y, Yim WWS, Yu F, Huang G . (2009). Late Quaternary environmental changes in the Pearl River mouth region, China. Quat Int 206: 35–45.
Acknowledgements
We thank Ms L.L. Ma, Mr W.W. Wong and Mr K.C. Cheung for their assistance in field collection and laboratory analysis and the Lands Department of the HKSAR Government for the preparation of and approval for using the map. The constructive comments from three anonymous reviewers have much improved the manuscript. We would also like to thank Dr D. Wilmshurst and Mr Zachary Chai for editorial comments on this manuscript. This study was supported by research grants (33/2009 and 28/2013) from the Environment and Conservation Fund, Government of Hong Kong Special Administrative Region, China.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article

Cite this article
Wong, W., Ma, K., Tsang, L. et al. Genetic legacy of tertiary climatic change: a case study of two freshwater loaches, Schistura fasciolata and Pseudogastromyzon myersi, in Hong Kong. Heredity 119, 360–370 (2017). https://doi.org/10.1038/hdy.2017.47
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2017.47
This article is cited by
-
Phylogeographic structure and population demography of the leopard mandarin fish (Siniperca scherzeri) in the Pearl River drainage
Environmental Biology of Fishes (2022)
-
Cryptic lineages and hybridization of the predaceous chub Parazacco spilurus (Actinopterygii, Cypriniformes, Xenocyprididae) in Hong Kong
Hydrobiologia (2019)
